Researchers overcome computational limitations to predict the starting materials of multi-step reactions using only information about the target product molecule.
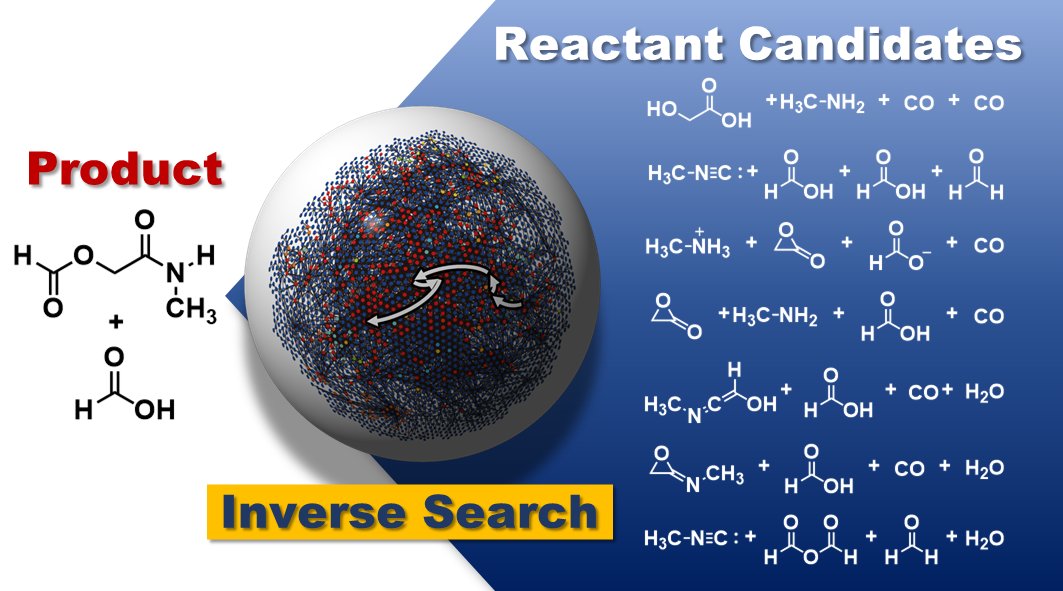
(Yosuke Sumiya, Yu Harabuchi, Yuuya Nagata, Satoshi Maeda. JACS Au. April 22, 2022)
Have you ever only caught the end of a TV show and wondered how the story progressed to that ending? In a similar way, chemists often have a desired molecule in mind and wonder what kind of reaction could produce it. Researchers in the Maeda Group at the Institute for Chemical Reaction Design and Discovery (ICReDD) and Hokkaido University developed a method that can predict the "story" (i.e., the starting materials and reaction paths) of multi-step chemical reactions using only information about the "ending" (i.e., the product molecules).
Predicting the recipe for a target product molecule, with no other knowledge than the molecule itself, would be a powerful tool for accelerating the discovery of new reactions. The Maeda group previously developed a computational method that succeeded in predicting single step reactions in this way. However, expanding to multi-step reactions leads to a dramatic increase in the number of possible reaction pathways-what is known as combinatorial explosion. This sharp increase in complexity results in prohibitively high calculation costs.
To overcome this limitation, researchers developed an algorithm that reduces the number of paths that need to be explored by discarding less viable paths at each step in the reaction. After calculating all possible paths for one step backward in the reaction, a kinetic analysis method evaluates how well each path produces the target molecule. Reaction paths that do not yield the target molecule above a pre-set threshold percentage are deemed not significant enough, and are not explored further.
This cycle of exploring, evaluating, and discarding reaction paths is repeated for each step backward in a multi-step reaction and mitigates the combinatorial explosion that would normally occur, making multi-step reactions more feasible to calculate. Previous methods were limited to single step reactions, whereas this new method was able to predict reactions that involved more than 6 steps, marking a major jump in capability.
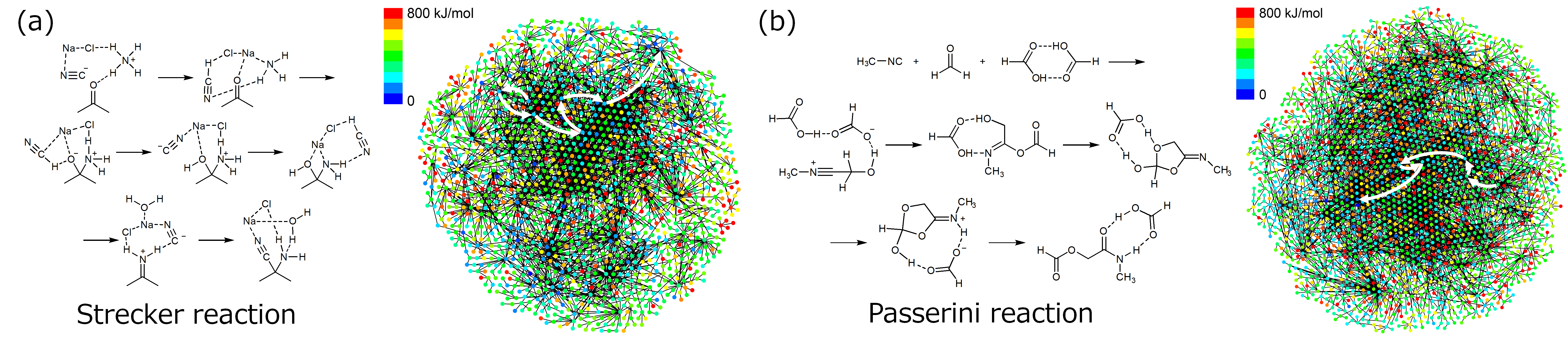
Generated structures and reaction path networks for the predicted reaction steps for the Strecker reaction (a) and Passerini reaction (b). White arrows show the multi-step path corresponding to the known reaction mechanism. (Image Credit: Satoshi Maeda)
As a proof-of-concept test, researchers tested the method on two well-known multi-step reactions, the Strecker and Passerini reactions. Thousands of starting material candidates were proposed for each reaction, which were filtered to the most promising candidates based on stability and product yield. Critically, among the proposed candidates were the well-known starting materials for each reaction, confirming the ability of the technique to identify experimentally viable starting materials from just the target product molecule.
Although further work is required to enable predicting even larger and more complex systems, researchers anticipate that this breakthrough in handling multi-step processes will accelerate the discovery of novel chemical reactions.
"This work provides a unique approach, as it is the first time performing reverse predictions of multi-step reactions using quantum chemical computations is possible without using any knowledge or data about the reaction," said Professor Satoshi Maeda. "We expect this technique will enable the discovery of entirely unimagined chemical transformations, in which case there is little knowledge or experimental data to use."

(Left to Right) Yosuke Sumiya, Yu Harabuchi, Yuuya Nagata, and Satoshi Maeda of the research team (Photo Credit: RIKEN and ICReDD).
Original Article:
Yosuke Sumiya, Yu Harabuchi, Yuuya Nagata, Satoshi Maeda. Quantum chemical calculations to trace back reaction paths for the prediction of reactants. JACS Au. Apr 22, 2022.
DOI: 10.1021/jacsau.2c00157
Funding:
This work was supported by the Japan Science and Technology Agency (JST) via Exploratory Research for Advanced Technology (ERATO) grant JPMJER1903. Support was also provided by the Institute for Chemical Reaction Design and Discovery (ICReDD), which was established by the World Premier International Research Initiative (WPI), Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan.





