A study from researchers at The University of Texas MD Anderson Cancer Center, published today in Nature Cell Biology, details a previously unexplained type of cell death called disulfidptosis that could open the door for novel cancer therapeutic strategies.
As described in the study, disulfidptosis is triggered when cells with high levels of the SLC7A11 protein are subjected to glucose starvation. In preclinical models, treatment with glucose inhibitors induced disulfidptosis in cancer cells with high SLC7A11 expression, effectively suppressing tumor growth without significant toxicity in normal tissues.
The study was led by Boyi Gan, Ph.D., and Junjie Chen, Ph.D., both professors of Experimental Radiation Oncology.
"Cancer cells rely on SLC7A11 to import cystine for maintaining redox balance and for cell survival. However, this also exposes an Achilles heel in SLC7A11-high cancer cells because these cells are dependent on glucose to resolve their disulfide-overloading issue," Gan said. "Starving these cells of glucose can overwhelm them with toxic disulfide molecules, resulting in rapid cell death."
Many cancers, such as lung cancer and kidney cancer, have an overexpression of SLC7A11, which codes for the cystine transporter. In a 2020 paper, Gan's team showed certain cancer cells might be susceptible to treatment with glucose transporter inhibitors due to their high expression of SLC7A11 and the resulting "addiction" to extracellular glucose.
The SLC7A11 protein imports cystine, an important amino acid that can contribute to tumor growth, but elevated levels of cystine and other disulfide molecules can be toxic. To regulate this balance, cells are forced to use the molecule NADPH to quickly convert toxic disulfides into other non-toxic molecules. NADPH is mainly supplied from glucose, so cutting off the glucose supply can lead to an accumulation of disulfide molecules and cell death.
The precise mechanism behind this process was not previously understood. According to Gan, this new study sheds light on the topic by demonstrating a previously uncharacterized form of cell death.
One of the best-known cell death mechanisms is apoptosis, which can be triggered either internally or externally, resulting in the activation of caspases which kill the cell by chopping up key proteins. Another highly studied cell death pathway in recent years is ferroptosis, which is caused by the accumulation of lipid peroxides.
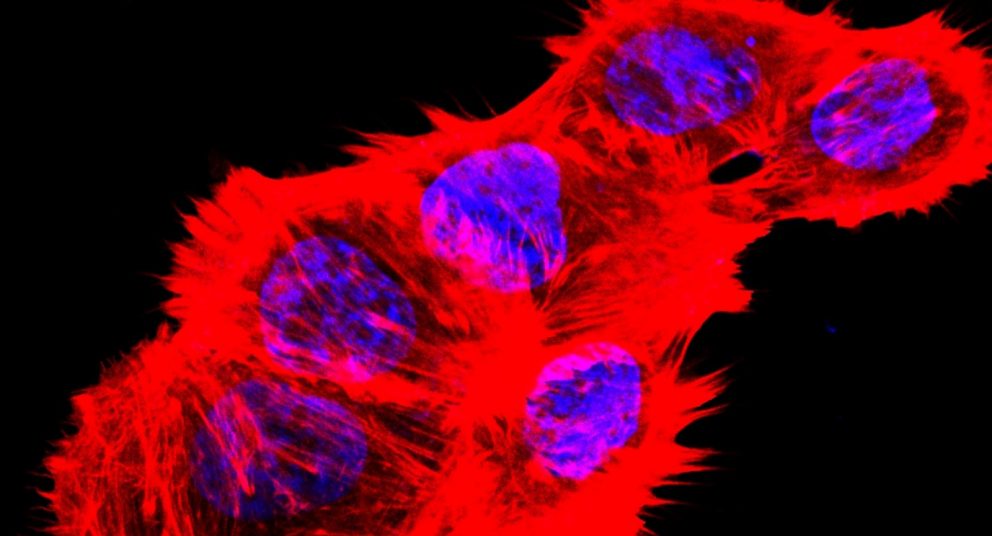
Disulfidptosis is different from these other cell death mechanisms because it relates to the actin cytoskeleton, a cell structure vital for maintaining cell shape and survival. The actin cytoskeleton is composed of actin filaments, which give cells their overall shape and structure.
This new study revealed that, in glucose-starved SLC7A11-high cancer cells, the large number of accumulated disulfide molecules cause aberrant disulfide bonding among actin cytoskeleton proteins, interfering with their organization and ultimately leading to actin network collapse and cell death.
Many cancer therapies are designed to kill cancer cells via apoptosis. However, many cancer cells find ways to escape from therapy-induced apoptosis, leading to therapy resistance and disease relapse. These findings suggest that targeting disulfidptosis merits further study as a cancer treatment approach.
"This important finding will hopefully inspire disulfidptosis-inducing treatments for cancers that have evaded other therapies and are resistant to apoptosis," Gan said. "Because SLC7A11 is highly expressed in many cancers, there might be a therapeutic window to inhibit glucose transporters and induce disulfidptosis in these cells while leaving normal cells unaffected."
According to Gan, the next direction of this research includes investigating how disulfidptosis can be initiated in other conditions and what additional pathways play a role in triggering it. Further understanding of these mechanisms could provide additional targets for cancer therapies.
This research was supported by the Institutional Research Fund and Bridge Fund from MD Anderson, the Emerson Collective, and the National Institutes of Health (R01CA181196, R01CA244144, R01CA247992, R01DK107733 and R35GM130119). A full list of authors and their disclosures can be found in the full paper here.