
Sooty mangabeys of West Africa host a strain of the simian immunodeficiency virus (SIV) that crossed over into humans sometime within the last 100 years and led to the spread of HIV-2, a less transmittable but equally deadly strain of the virus. (iStock image)
For hundreds of thousands of years, monkeys and apes have been plagued by simian immunodeficiency virus (SIV), which still devastates primate groups in Africa.
Luckily, as humans evolved from these early primates, we picked up a mutation that made us immune from SIV - at least until the early 20th century, when the virus evolved to get around our defenses, giving rise to human immunodeficiency virus (HIV) and an AIDS pandemic that today affects an estimated 38 million people worldwide.
University of California, Berkeley, researchers have now discovered how that long-ago human mutation interfered with SIV infection, a finding that could provide clues for the development of new therapies to thwart HIV and similar viral infections.
"The main importance for this paper is that it tells us what was one of the last major barriers before the crossover to humans happened," said James Hurley, a UC Berkeley professor of molecular and cell biology. "The current paper is an archeological look at how this happened."
The barrier was a mutation in human cells that blocked SIV from forcing these cells to shed thousands more copies of the virus. As a result, humans could not re-infect one another.
This genetic mutation interfered with the ability of an SIV protein to tightly bind two human proteins and send them for destruction within the cell, instead of fighting the virus. The researchers used cryo-electron microscopy, or cryoEM, to determine the structure of this protein complex and discovered that the mutation so effectively disrupted the protein binding sites that it took SIV a long time to find a work-around.
"The binding site involved is structurally very complex, so essentially it is not possible to adapt to it once the tight binding is lost. The virus had to invent a completely different way to do the same thing, which took a long time in evolution," Hurley said. "This conferred an advantage on our prehistoric ancestors: From chimps on down, every primate was susceptible to SIV, but humans were immune. That gave humans probably a grace period of tens to hundreds of thousands of years to develop without having to deal with this disease. I tend to think that really gave a leg up to humans in early evolution."
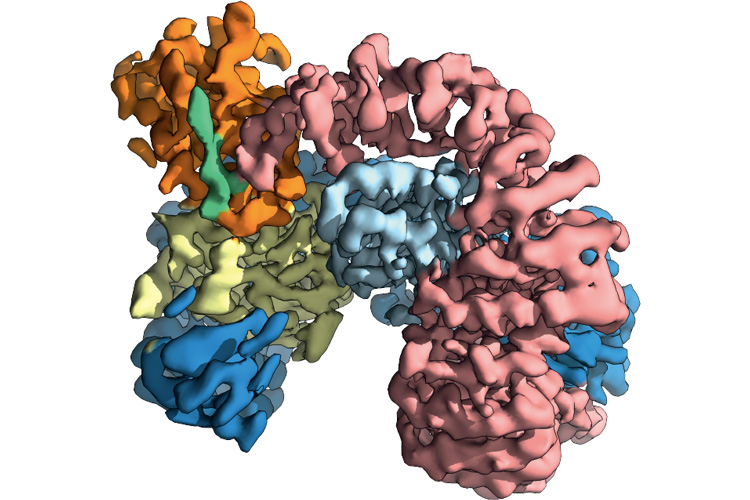
A cryoEM image of the tightly bound, three-protein complex of tetherin (green), the SIV protein Nef (orange) and the primate protein AP-2 (other colors). By forming this complex, SIV forced monkey and ape host cells to destroy tetherin, leaving SIV free to multiply by the thousands. A mutation in human tetherin disrupted this binding, leaving early humans free from SIV infection for millennia. (UC Berkeley image courtesy of James Hurley)
Though the SIV virus - in this case, from a monkey called the sooty mangabey (Cercocebus atys), the source of the less virulent HIV-2 strain in humans - differs in several ways from the HIV strains that afflict humans, the findings could pinpoint targets for drugs as researchers look for "functional" cures for AIDS. These would be one-time treatments that prevent flare-ups of the disease, even if the virus remains in the body.
"The overall strategy in our lab is to try to find regions in the structures of human proteins that are attacked by viruses, but are not needed for normal purposes by the host, so that a drug can be designed to attack that region," Hurley said. "The virus will typically respond by mutating, which means it evolves drug resistance, but this new finding suggests that with the right point of attack, it could take SIV or HIV, in some cases, tens of thousands of years of evolution to catch up."
The work will be published in the Sept. 11 issue of the journal Cell Host & Microbe and was posted online Aug. 22.
Sooty mangabeys
SIV and HIV, which are lentiviruses, are hard to root out the body because they insert their DNA into the genomes of host cells, where it sits like a ticking time bomb, ready at any moment to revive, take over the host cell's machinery to makes copies of itself and send out thousands of these copies - called virions - to infect other cells.

The tetherin protein (green) on the surface of monkey, ape and human cells inhibits the release of SIV virions from the cell (left). SIV overcomes this restriction by expressing the protein Nef (yellow), which down-regulates tetherin by tying it to the protein AP-2 (purple). This tight Nef binding is absent in humans. The inability of SIV to destroy human tetherin was one of the major barriers to crossover of SIV to humans. (UC Berkeley image courtesy of Cosmo Buffalo)
These virions are formed when the newly copied viral DNA wraps itself in a piece of the host cell's membrane and buds off, safely ensconced in a bubble until it can reinfect.
Because budding is an important step in the spread of many viruses, primates long ago evolved natural defenses, including proteins on the surface of cells that staple the budding virions to the cell and prevent them from leaving. As they accumulate, the immune system recognizes these unbudded virions as abnormal and destroys the whole cell, virus and all.
In monkey, ape and human cells, the staple is called tetherin, because it tethers the budding virion to the cell membrane.
In the constant arms race between host and pathogen, SIV evolved a countermeasure that exploits another normal cell function: its recycling system. Cells have ways to remove proteins sitting on the surface, through which cells constantly take up and recycle tetherin if there's no indication it is needed to fight an invading virus. It does this by dimpling the membrane inward to form a little bubble inside the cell, capturing tetherin and other surface proteins in this vesicle and then digesting all the contents, including tetherin.
SIV's countermeasure was to produce a protein, called Nef, that revs up the recycling of tetherin, even during an infection. This enables virions to bud off and search for new victims.
Hurley and project scientist Xuefeng "Snow" Ren found that Nef forms a tight wedge between tetherin and a protein in the vesicle called AP-2, preventing tetherin from escaping the vesicle and dooming it to recycling.
"Nef is a bridge between AP2 and tetherin to recruit them into endocytosis, dragging the tetherin into the vesicle," Ren said. "So it tricks our own cells' machinery for getting rid of stuff we don't want into getting rid of stuff the virus doesn't want."
The five amino acids that humans lost in the tetherin protein - the mutation that gave humans immunity against SIV - loosened the binding between tetherin, Nef and AP-2, which allowed tetherin to escape recycling. This blocked the crossover of zoonotic virus transmission, Ren said, because the structural rearrangement was so extensive that SIV couldn't fix it by simple mutations in Nef.
SIV developed a new trick
Some variants of SIV did eventually find a way around this hurdle, however. At some point, a few SIVs acquired a second protein, Vpu, to do what Nef also did - wedge itself between proteins to cement connections helpful to the virus. At some point, perhaps a hundred years ago, this strain of SIV moved into humans from chimpanzees, and a slight mutation in Vpu reignited the recycling of tetherin in humans, unleashing what we know today as group M HIV-1, the most virulent form of HIV worldwide.
"There were probably many crossovers into humans that failed, but eventually, some hunter in Africa, perhaps in the course of butchering a chimp, was exposed to the blood, and the virus then acquired an additional mutation, a small step that turned SIV into HIV," Hurley said.
Next up, Hurley, Ren and their colleagues plan to use cryoEM to determine the structure of the three-protein complex in gorilla variants of SIV, which evolved into the O strain of HIV-1, a less virulent strain that originated in the African country of Cameroon.
Other co-authors of the paper are first author and UC Berkeley postdoctoral fellow Cosmo Buffalo and Christina Stürzel, Elena Heusinger, Dorota Kmiec and Frank Kirchhoff of the University of Ulm in Germany, who used tests of viral infectivity to confirm that the explanation coming from cryoEM was correct in both monkey and human cells.
This research was supported by the National Institutes of Health (R01 AI120691, P50 GM082250, F32 GM125209) and the Andrew Dougherty Vision Foundation.





