Scientists at the University of North Carolina at Chapel Hill School of Medicine and colleagues have demonstrated that variants in the SPTBN1 gene can alter neuronal architecture, dramatically affecting their function and leading to a rare, newly defined neurodevelopmental syndrome in children.
Damaris Lorenzo, PhD, assistant professor in the UNC Department of Cell Biology and member of the UNC Neuroscience Center at the UNC School of Medicine, led this research, which was published today in the journal Nature Genetics. Lorenzo, who is also a member of the UNC Intellectual and Developmental Disabilities Research Center (IDDRC) at the UNC School of Medicine, is the senior author.
The gene SPTBN1 instructs neurons and other cell types how to make βII-spectrin, a protein with multiple functions in the nervous system. Children carrying these variants can suffer from speech and motor delays, as well as intellectual disability. Some patients have received additional diagnosis, such as autism spectrum disorder, ADHD, and epilepsy. Identification of the genetic variants that cause this broad spectrum of disabilities is the first important milestone to finding treatments for this syndrome.
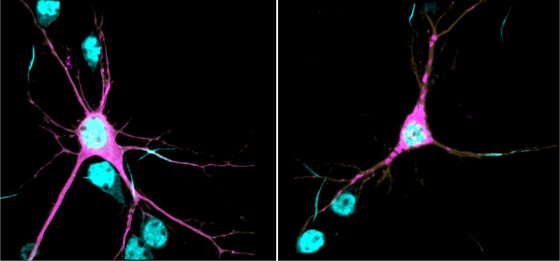
Right, βII-spectrin (magenta) forms aggregates throughout neurites of a mouse cortical neuron expressing one of the human SPTBN1 variants. (Lorenzo Lab)
Lorenzo first learned about patients with complex neurodevelopmental presentations carrying SPTBN1 variants from Queenie Tan, MD, PhD, a medical geneticist, and Becky Spillmann, MS, a genetic counselor - both members of the NIH-funded Undiagnosed Disease Network (UDN) site at Duke University and co-authors of the Nature Genetics paper. They connected with Margot Cousin, PhD, a geneticist associated with the UDN site at the Mayo Clinic and co-first author or the study. Cousin had also collected clinical information from SPTBN1 variant carriers. Other clinical genetics teams learned about these efforts and joined the study.
The cohort of individuals affected by SPTBN1 variants continues to grow. Lorenzo and colleagues have been contacted about new cases after they published a preprint of their initial findings last summer. Identifying the genetic cause of rare diseases such as the SPTBN1 syndrome requires pooling knowledge from several patients to establish common clinical and biological patterns.
"Fortunately, the advent of affordable gene sequencing technology, together with the creation of databases and networks to facilitate the sharing of information among clinicians and investigators, has vastly accelerated the diagnosis of rare diseases," Lorenzo said. "To put our case in historical perspective, βII-spectrin was co-discovered 40 years ago through pioneering work that involved my UNC colleagues Keith Burridge, PhD, and Richard Cheney, PhD, as well as my postdoctoral mentor Vann Bennett, PhD, at Duke. However, its association with disease eluded us until now."
βII-spectrin is tightly associated with the neuronal cytoskeleton - a complex network of filamentous proteins that spans the neuron and plays pivotal roles in their growth, shape, and plasticity. βII-spectrin forms an extended scaffolding network that provides mechanical integrity to membranes and helps to orchestrate the correct positioning of molecular complexes throughout the neuron. Through research published in PNAS in 2019, Lorenzo found that βII-spectrin is essential for normal brain wiring in mice and for proper transport of organelles and vesicles in axons - the long extensions that carry signals from neurons to other neurons. βII-spectrin is an integral part of the process that enables normal development, maintenance, and function of neurons.
In this new study, Lorenzo's research team showed that, at the biochemical level, the genetic variants identified in patients are sufficient to cause protein aggregation, aberrant association of βII-spectrin with the cytoskeleton, impair axonal organelle transport and growth, and change the morphology of neurons. These deficiencies can permanently alter how neurons connect and communicate with each other, which is thought to contribute to the etiology of neurodevelopmental disorders. The team showed that reduction of βII-spectrin levels only in neurons disrupts structural connectivity between cortical areas in mutant mice, a deficit also observed in brain MRIs of some patients.
In collaboration with Sheryl Moy, PhD, professor in the UNC Department of Psychiatry and director of the Mouse Behavioral Phenotyping (MBP) Core of the UNC IDDRC, the researchers found that these mice have developmental and behavioral deficits consistent with presentations observed in humans.
"Now that we've established the methods to assign likelihood of pathogenicity to SPTBN1 variants and to determine how they alter neurons, our immediate goal is to learn more about the affected molecular and cellular mechanisms and brain circuits, and evaluate strategies for potential clinical interventions," Lorenzo said.
To this end, her team will collaborate with Adriana Beltran, PhD, assistant professor in the UNC Department of Genetics and director of the UNC Human Pluripotent Cell Core, to use neurons differentiated from patient-derived induced pluripotent stem cells. And the research team will continue to tap into molecular modeling predictions in collaboration with Brenda Temple, PhD, professor in the UNC Department of Biochemistry and Biophysics and director of the UNC Structural Bioinformatics Core, both co-authors on the Nature Genetics paper.
"As a basic science investigator, it's so satisfying to use knowledge and tools to provide answers to patients," Lorenzo said. "I first witnessed this thrill of scientific discovery and collaborative work as a graduate student 15 years ago when our lab identified the genetic cause of the first spectrinopathy affecting the nervous system, and it has been a powerful motivator since."
That work was the discovery of variants in a different spectrin gene as the cause of spinocebellar ataxia type 5 (SCA5), led by Laura Ranum, PhD, who at the time was at the University of Minnesota. In follow up work, as part of that team, Lorenzo contributed insights into the pathogenic mechanism of SCA5.
"Aside from the immediate relevance to affected patients, insights from our work on SPTNB1 syndrome will inform discoveries in other complex disorders with overlapping pathologies," Lorenzo said. "It is exciting to be part of such important work with a team of dedicated scientists and clinicians."
Members of the Lorenzo lab who are co-authors in the Nature Genetics paper are co-first author Blake Creighton, lab research technician in the Lorenzo lab; Reggie Edwards, graduate student; Keith Breau, graduate student at the time of this research; Deepa Ajit, PhD, a postdoctoral fellow; Sruthi Dontu, Simone Afriyie, and Julia Bay, all undergraduates at UNC-Chapel Hill; and Liset Falcon, lab research technician at the time of this research. Other UNC-Chapel Hill collaborators and co-authors in the paper are Kathryn Harper, PhD, project manager in the MBP Core; and Lorena Munoz and Alvaro Beltran, both research associates in the hHPSC.
This research was funded by grants from the National Institutes of Health and the National Ataxia Foundation.





