A research team has developed a unified theoretical framework to better predict the performance of single-atom catalysts (SACs) for electrochemical carbon dioxide reduction (CO₂RR). Their model incorporates both pH and interfacial electric field effects - two critical factors that have often been overlooked or oversimplified in conventional catalyst studies.
CO₂RR is gaining attention as a potential solution for reducing greenhouse gas emissions by converting carbon dioxide into useful products under mild conditions. Among the various outputs, carbon monoxide (CO) is especially valuable due to its use in syngas mixtures and chemical synthesis. However, catalyst behavior during CO₂RR is known to be highly sensitive to the surrounding electrolyte environment, particularly local pH levels.
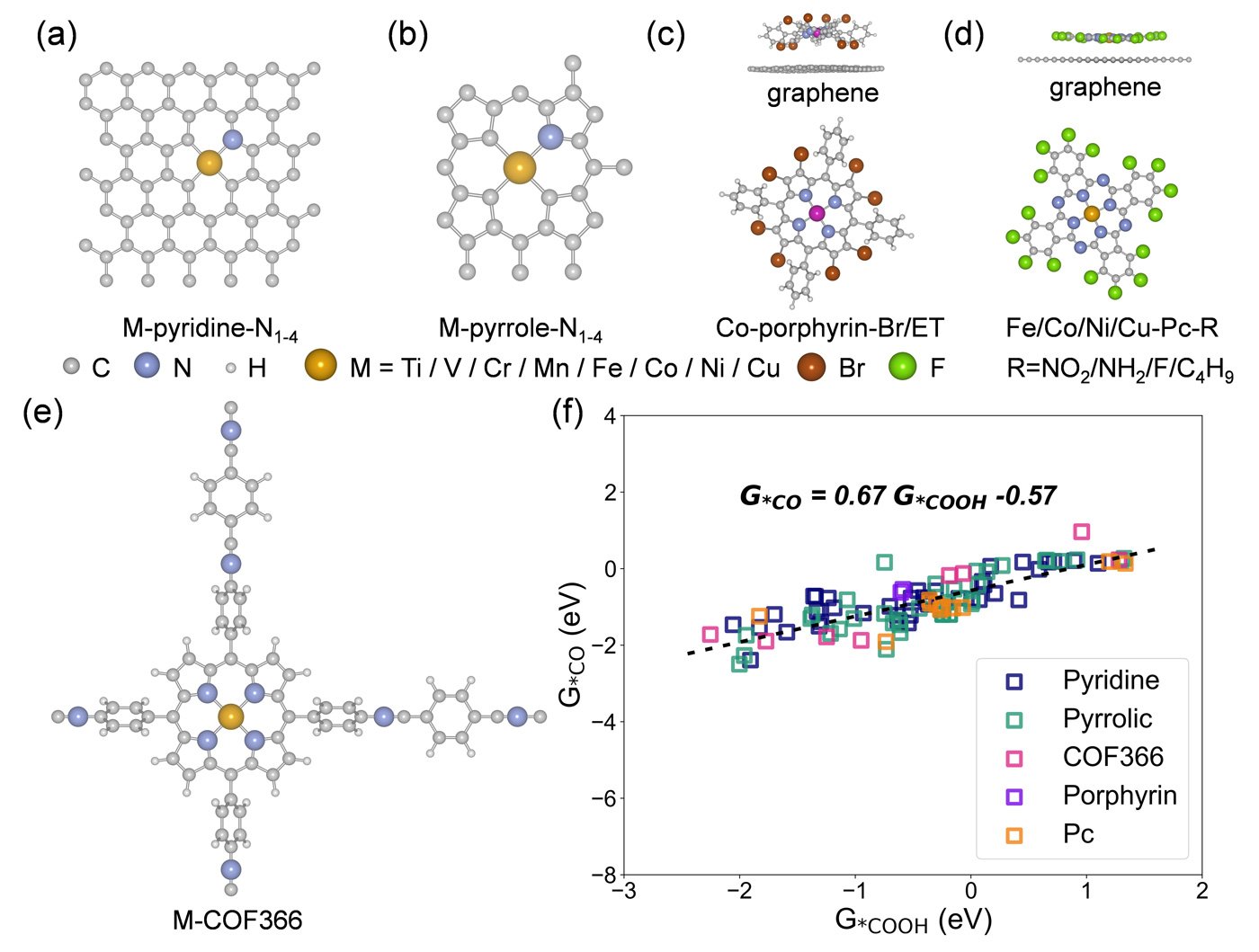
To address this, the team created a microkinetic model that operates at the reversible hydrogen electrode (RHE) scale. It explicitly accounts for the dynamic influence of pH and electric fields on reaction intermediates. This model was combined with spin-polarized density functional theory (DFT-D3) calculations and data-driven screening to evaluate 101 SAC configurations based on d-block transition metals.
The study examined SACs across a range of coordination environments, including pyrrole- and pyridine-functionalized graphene, covalent organic frameworks (COFs), porphyrins, and phthalocyanines. A consistent linear relationship was found between the adsorption energies of the key intermediates - ∗COOH and ∗CO - indicating a predictable trend in binding behavior.
One of the notable findings of the study was the role of dipole-field interactions in modulating these intermediate bindings under varying pH conditions. These interactions helped explain why certain catalysts perform differently across pH environments, offering deeper insight into the mechanisms at play in CO₂RR.
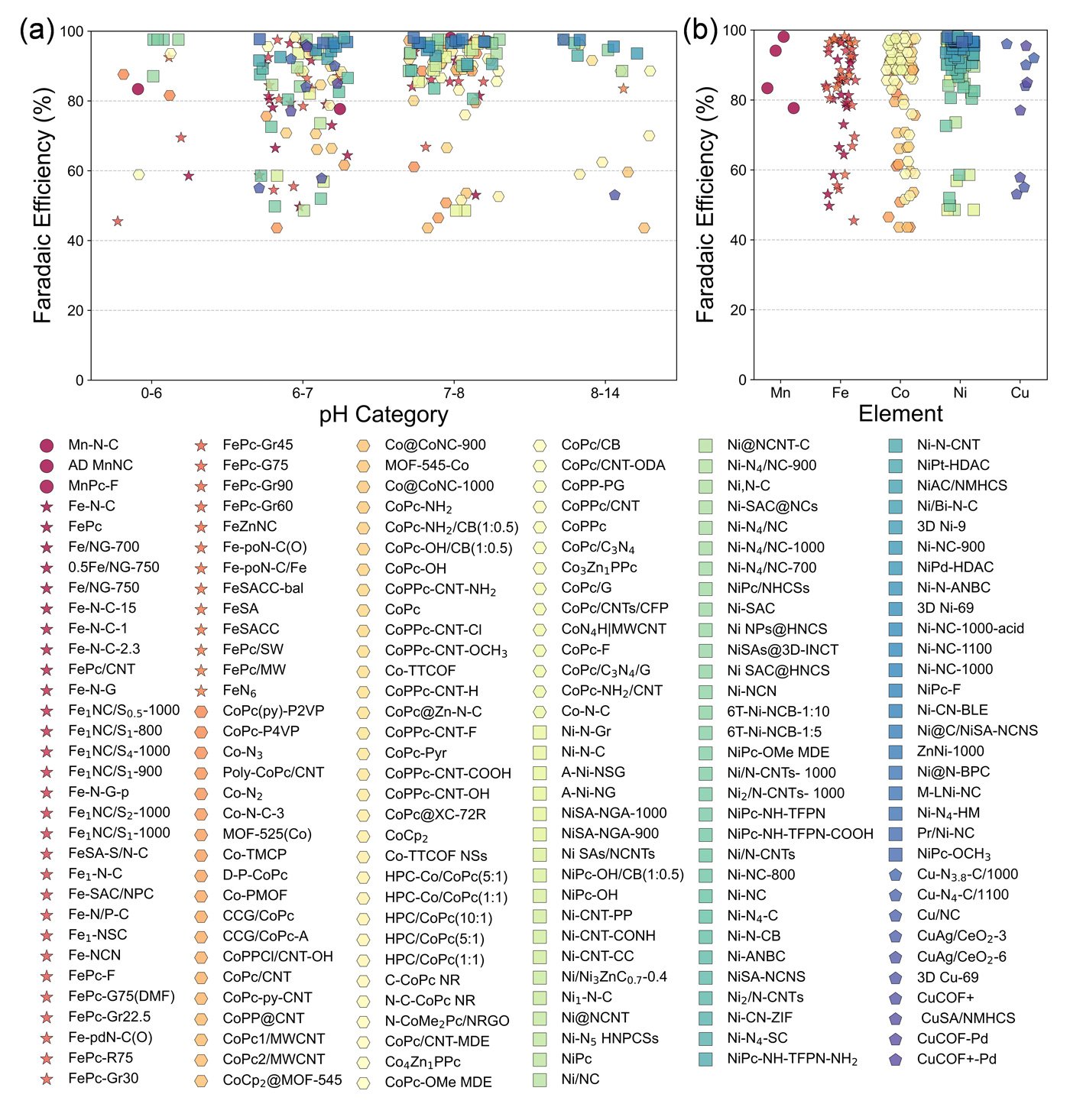
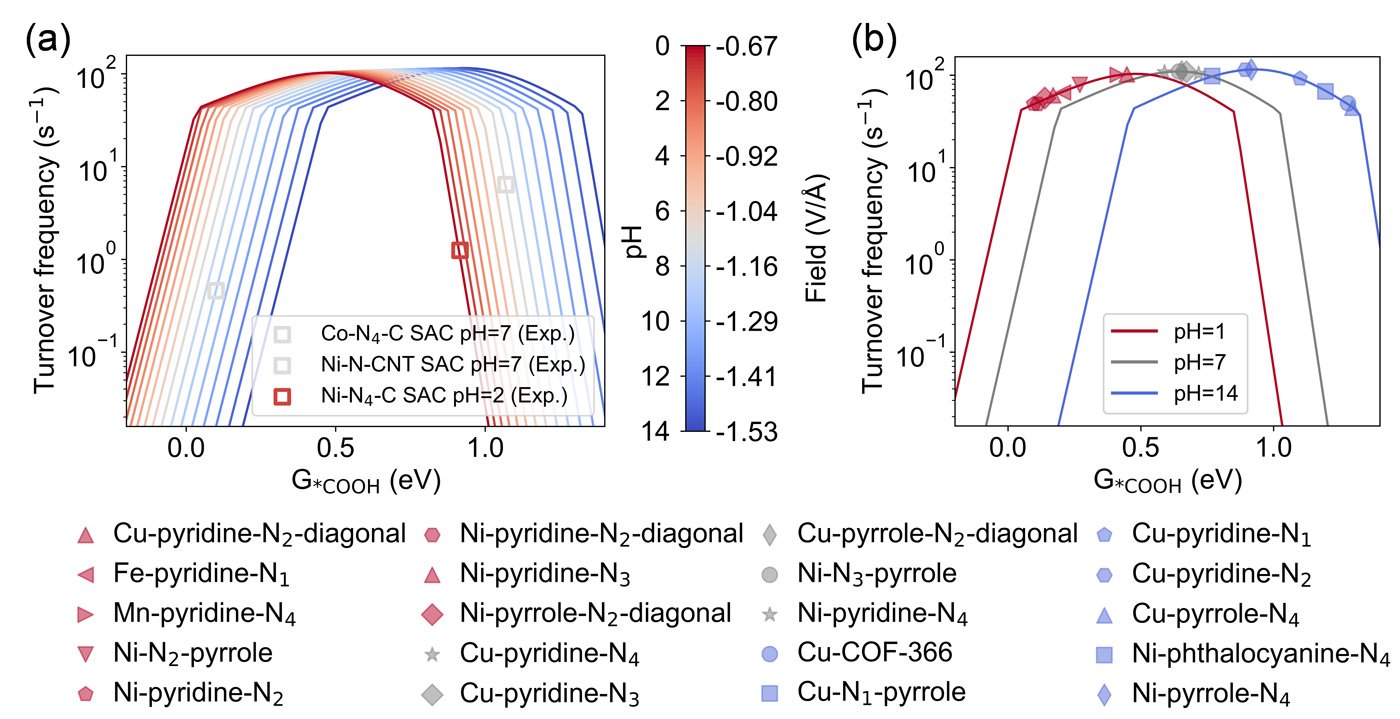
Model predictions showed strong agreement with available experimental turnover frequency data, suggesting the framework's reliability in real-world applications. Based on this agreement, the researchers identified 12 SACs with favorable CO selectivity across a wide range of pH values. These catalysts are primarily centered around iron (Fe), copper (Cu), and nickel (Ni) atoms.
"This work represents a step toward more predictive catalyst screening by bridging theory with realistic electrochemical conditions," said Hao Li, Professor at Tohoku University's Advanced Institute for Materials Research (WPI-AIMR) and lead author of the study. "By integrating pH and field effects into our model, we can more accurately guide the design of catalysts suited for carbon-neutral energy technologies."
All computational structures used in the study have been uploaded to the Digital Catalysis Platform, a growing open-access resource for the catalysis community developed by Professor Li's group. This contribution aims to support further research and data sharing across the field.
Looking ahead, the team plans to refine the model further to improve both predictive accuracy and computational efficiency. They also aim to incorporate machine learning algorithms to accelerate catalyst design and deepen understanding of pH-sensitive electrochemical reactions.
- Publication Details:
Title: Data-driven discovery of single-atom catalysts for CO2 reduction considering the pH-dependency at the reversible hydrogen electrode scale
Authors: Yue Chu, Yuhang Wang, Di Zhang, Xuedan Song, Chang Yu, and Hao Li
Journal: The Journal of Chemical Physics
DOI: 10.1063/5.0267969





