Protein dynamics play a crucial role in their diverse functions. The intracellular environment significantly influences protein dynamics, particularly for intrinsically disordered proteins (IDPs).
A research group led Prof. ZHANG Lihua from the Dalian Institute of Chemical Physics (DICP) of the Chinese Academy of Sciences (CAS), in collaboration with Assoc. Prof. GONG Zhou from the Precision Measurement Science and Technology Innovation Research Institute of CAS, has proposed a strategy using in-vivo chemical cross-linking and mass spectrometry (in-vivo XL-MS) to decode the dynamic structure of proteins within cells.
In-vivo XL-MS is potential for analyzing the dynamic structure of proteins within cells due to its high throughput, high sensitivity, and low requirements for protein purity.
This study was published in Angewandte Chemie International Edition on July 5.
AlphaFold, a previously proposed protein structure prediction method, combines novel neural network architectures and training procedures that are based on the evolution of structural patterns and the integration of physical and geometric restraints.
In this study, the researchers utilized the prior structure obtained from AlphaFold2 as prior information, and combined in-vivo XL-MS data with various structural calculation methods to assess the compatibility between structures and cross-linking information. They reconstructed the dynamic structures of various proteins within cells.
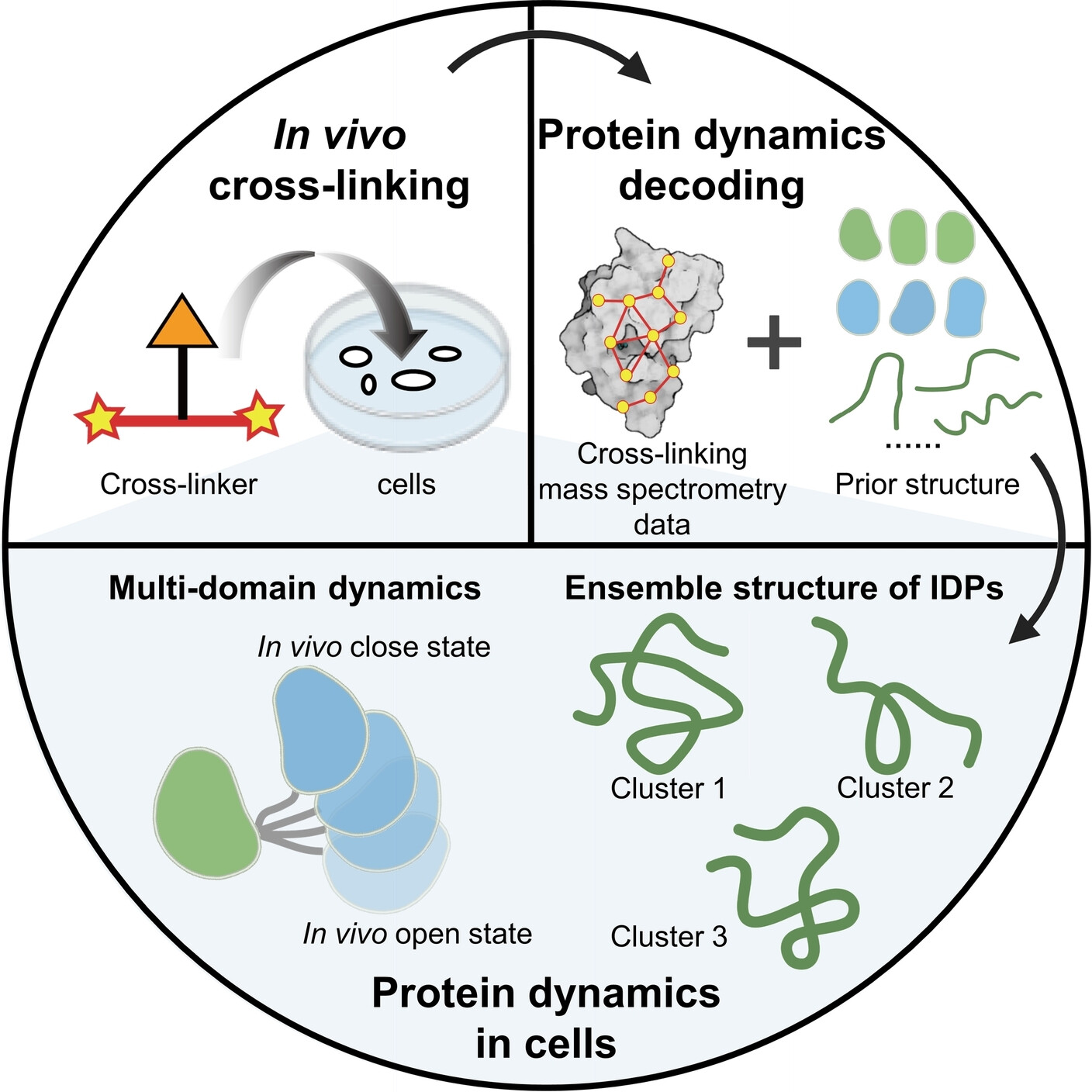
They focused on multi-domain proteins and proposed a strategy to treat the domains as a whole, utilizing XL-MS data between the domains to model the dynamic structures of proteins within cells. They characterized the dynamic structures of three multi-domain proteins, namely calmodulin, hnRNP A1, and hnRNP D0.
As for IDPs, they introduced two complementary structural characterization strategies: one involved directly converting XL-MS data into distance constraints for IDP structure calculation, while the other strategy employed unbiased sampling using all-atom molecular dynamics simulations, followed by evaluation and selection of the sampled structures based on XL-MS data. By employing these two strategies, they decoded the ensemble conformations of two highly mobile high-mobility group proteins within cells, HMG-I/Y and HMG-17.
"Our study provides technical support for a deeper understanding of the molecular mechanisms underlying protein functionality in the cellular microenvironment," said Prof. ZHANG.





