EMBL scientists co-initiated the reconstruction of the most diverse set of reference human genomes ever assembled
Credit: David Porubsky/University of Washington
In 2001, the International Human Genome Sequencing Consortium announced the first draft of the human genome reference sequence. The Human Genome Project had taken more than 11 years of work and involved more than 1,000 scientists from 40 countries. The reference sequence, however, does not represent a single individual but instead is a composite of genomes from several individuals that cannot accurately capture the complexity of human genetic variation.
Building on this, scientists have carried out many sequencing projects over the past 20 years to identify and catalogue genetic differences between individual genomes and the reference genome. These differences usually focused on small changes of a single letter of the DNA code and missed larger genetic alterations. Current technologies are now beginning to detect and characterise larger differences - called structural variants - such as insertions of several hundred letters. Structural variants are more likely than smaller genetic differences to interfere with gene function.
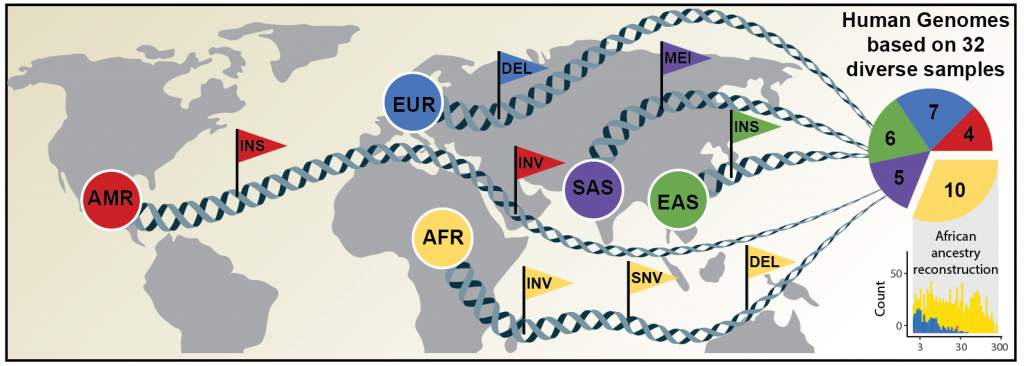
EMBL's Korbel group, in partnership with Heinrich Heine University Düsseldorf, The Jackson Laboratory in Farmington, Connecticut, and the University of Washington in Seattle, has now published an article in Science announcing a new, significantly more comprehensive reference dataset obtained using a combination of advanced sequencing and mapping technologies. The new reference dataset contains 64 assembled human genomes, representing 25 human populations from Africa, North America, East and South Asia, and Europe.
This study builds on a new method published by the researchers last year in Nature Biotechnology to accurately reconstruct the two components of a person's genome - one inherited from each of their parents. When assembling a person's genome, this method relied on a technology provided by EMBL known as Strand-seq to distinguish maternal and paternal DNA sequences.
"For each human individual that participated in the study, we identified not one but two genomes - one for each set of chromosomes," explains Jan Korbel, who led the research at EMBL. "Humans have two sets of chromosomes, which they receive from their parents. Previously we could not distinguish whether genetic variation came from one chromosome set or the other. We have now been able to solve this thanks to advances made by the Human Genome Structural Variation Consortium. It represents a remarkable achievement for the discovery of genetic variation in humans, which can now be studied much more comprehensively, leading the way to better find disease-causing genes."
The distribution of genetic variants can differ substantially between population groups as a result of spontaneous and continuously occurring changes in the genetic material. If such a mutation is passed on over many generations, it can become a genetic variant specific to that population.
The new reference data provide an important basis for including the full spectrum of genetic variants in genome-wide association studies, which examine genetic variants across the whole genome to find out whether any variants are associated with specific traits or diseases. The aim is to estimate an individual's risk of developing diseases such as cancer, and to understand the underlying molecular mechanisms. This, in turn, can be used as a basis for more targeted therapies and preventive medicine.
This work might enable further applications in precision medicine. Drug efficacy, for example, can vary between individuals based on their genomes. The new reference data now represent the full range of genetic variant types and incorporate human genomes of great diversity. "These genomes will enable a new wave of scientific discoveries about the biology of the human genome and the connection between genetic variation and disease," says EMBL researcher and co-first author Bernardo Rodriguez-Martin. "As an example, we were able to estimate the age of highly mutagenic L1 repeats. Very surprisingly, although these sequences originated up to three million years ago, they continue to mutate the human genome frequently, which occasionally leads to diseases such as cancer." This new resource might therefore contribute to developing novel approaches in personalised medicine, where the selection of therapies is tailored to a patient's individual genetic background.





