The number of known proteins is infinitely small in comparison to the universe of possible proteins which could in theory be realized. Yet these known proteins are the only major training ground for future protein design. Understanding how representative these proteins are of the overall potential diversity can therefore help inform strategies for a wide range of applications, including therapeutic, biocatalysis, or biomaterials development.
Published in PNAS, an international team from the Okinawa Institute of Science and Technology (OIST), the Institute of Science and Technology Austria (ISTA), the University of Vienna and the Centro de Astrobiología (CAB) investigated the relationship between protein evolution and sequence space, identifying the limiting factors behind protein diversification. Their findings reinforce theories of DNA recombination as a driving force of ancestral protein formation and highlight the limitations of many cutting-edge AI protein design methods.
"Modern AI methods are thought to be revolutionizing protein design, with the 2024 Nobel Prize in Chemistry awarded to the team behind AlphaFold. Yet most of these AI design methods are typically trained on databases of known proteins. So without understanding how representative these known proteins are of sequence space, how confident can we be that such methods can generate truly diverse protein designs?" says Professor Fyodor Kondrashov, head of OIST's Evolutionary and Synthetic Biology Unit.
Exploring the protein universe
Imagine you have 20 or so different block types, which you can connect in different orders and abundances into chains of tens, hundreds or even thousands of blocks in length. Mapping all possible resulting chains creates a sequence space.
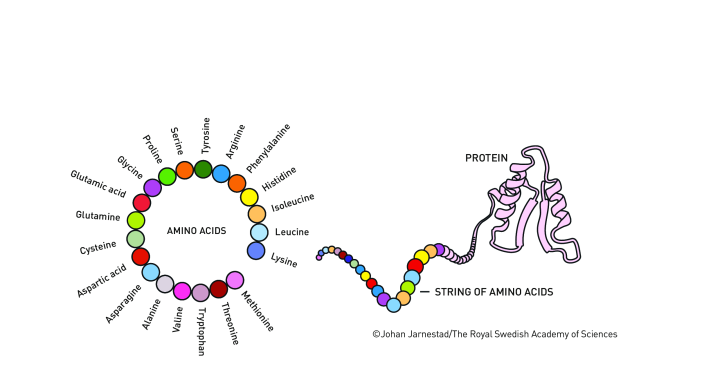
For proteins, the shape and structure of their amino acid building blocks mean only a minute fraction of possible protein sequences can fold up into the correct 3D shape to power a biological function. They need the correct chemical groups in the correct places to create the interactions that will maintain 3D shape or bind to other molecules. Mapping the sequences that fulfil this requirement creates a smaller functional space.
Of these possible functional sequences, it's likely that relatively few have ever existed across evolutionary history. Therefore, the researchers set out to uncover how representative this subset of proteins is of functional space.
The researchers started by mathematically describing the sequence space taken up by known proteins. They then built a model of protein evolutions to understand the biological factors controlling the structural diversification of a wide range of naturally-occurring protein families. From their models, they then predicted how many functional sequences they would expect to exist for a given biological function.
By comparing the diversity of known proteins to these theoretical predictions of protein evolution, the researchers found that point-of-origin effects far outweighed the influence of other key evolutionary processes.
"That starting point is the main evolutionary limit is not necessarily surprising, but the scale of its importance is really quite remarkable," observes lead author Lada Isakova, PhD student within the unit. "As an evolutionary biologist, I was intrigued to see how little selection and epistasis seemed to matter in our results."

What limits protein evolution?
When mutations arise in the genes encoding for a particular protein, these can result in changes to the sequence of amino acids produced, causing protein evolution. Natural selection limits which mutations persist over time based on whether they improve or harm the protein's function or stability. Epistasis - genetic interactions resulting in different outputs - also constrains evolution, as mutations may have limited effects alone, but large effects when present in combination with certain other mutations.
Both selection and epistasis are known to influence protein evolution, yet Isakova and colleagues found that by far, the limiting factor of protein diversity is the origins of our proteins, with relatively small divergence seen from the areas of sequence space of ancestral proteins.
This research provides new insights into the origins of life, reinforcing existing theories on initial protein formation. Isakova explains, "Our simulations suggest that, for the first proteins in the last universal common ancestor to arise, they couldn't just diverge from mutations of a single first sequence, given the time constraints we see. Instead, small pieces of DNA must have shuffled around and recombined to create new DNA molecules which could encode very different proteins."
The team also hopes that the research inspires experimental scientists to expand the known sequence space. Isakova comments, "Neural network approaches for functional protein prediction are limited by the data sets we provide. So based on existing data, most methods won't be able to generalize well beyond the current known sequence space. We can see there's huge swaths of sequence space left to be explored, but it'll take new experimental data to enable expansion into these unknown realms."
This global collaboration was supported by a Japan Science and Technology Agency (JST) Adopting Sustainable Partnerships for Innovative Research Ecosystem (ASPIRE) grant, which aims to build a network between top researchers in Japan and around the world, nurturing future scientific leaders.





