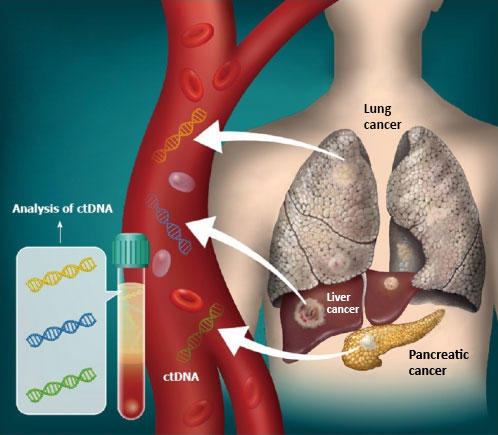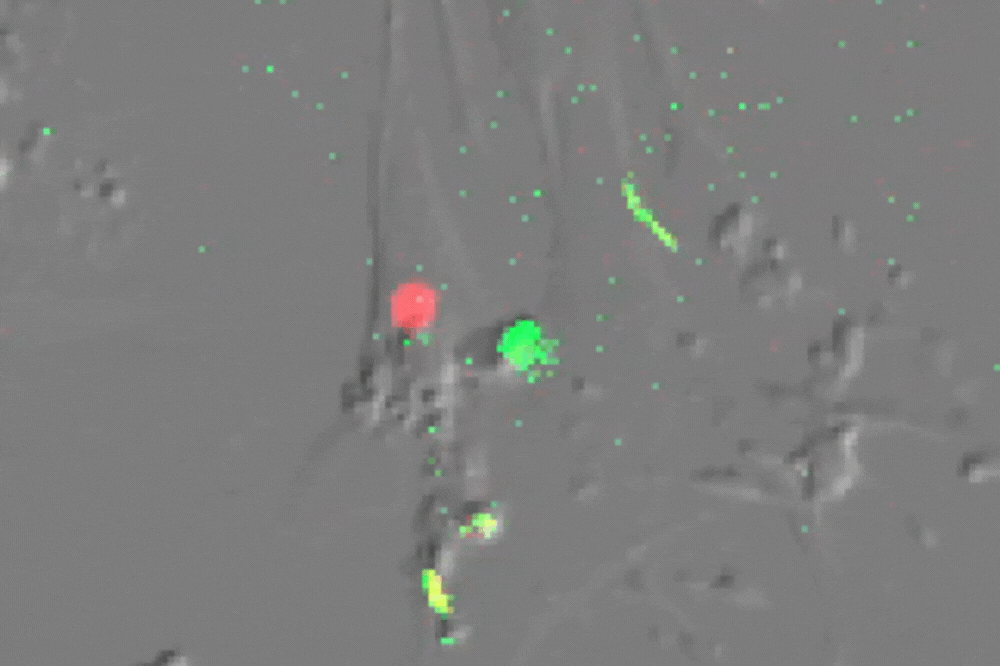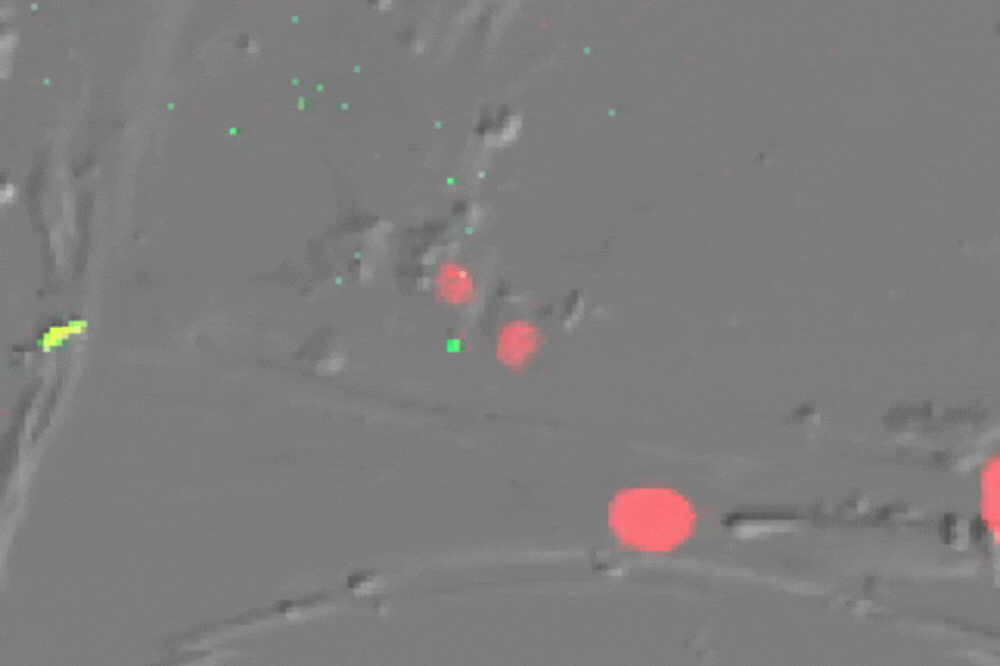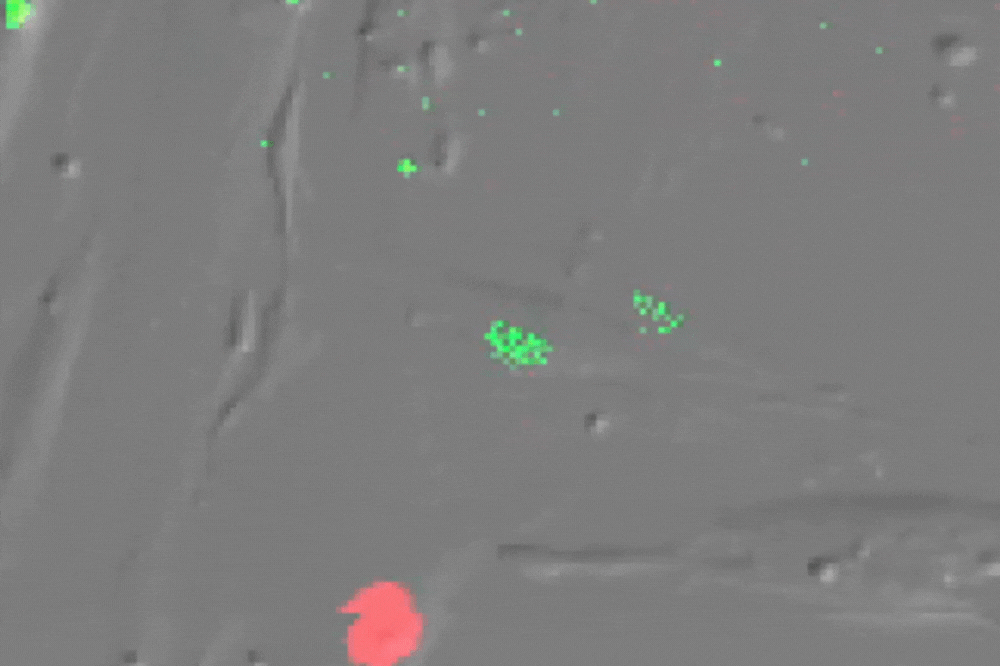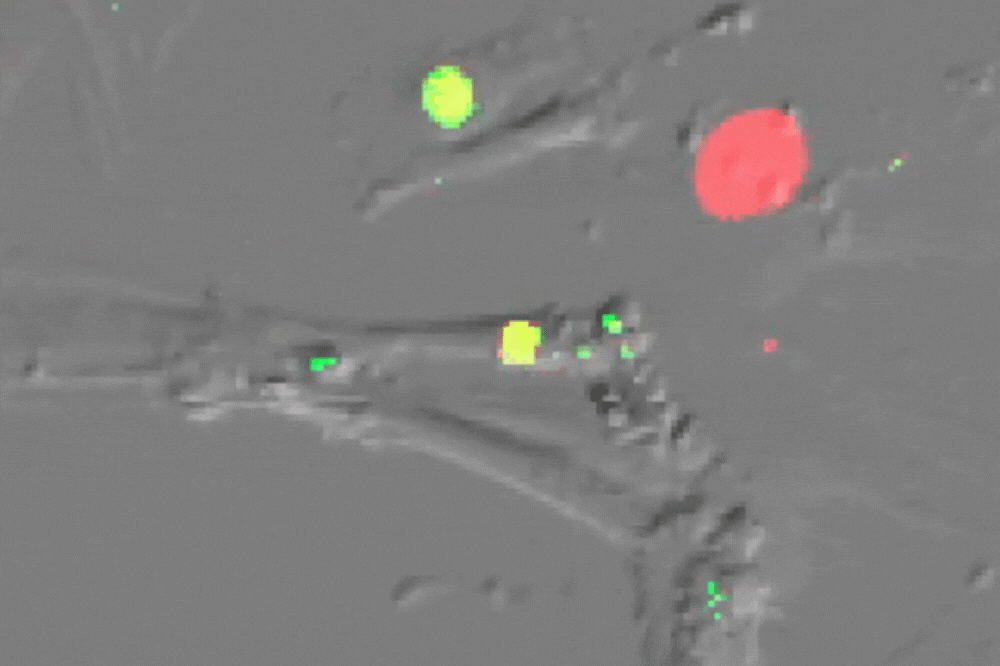
Single cell imaging of senescent cells (which usually do not divide) undergoing multiple divisions after ATM inhibition-their red nucleus turns green before division. (Credit: de Lange lab)
After a finite number of divisions, cells simply give up. As each round of replication trims their telomeres-the protective caps at the chromosome ends-those caps eventually become too short to prevent chromosome ends from being recognized as DNA breaks. As a result, cells permanently arrest, a phenomenon known as replicative senescence. This telomere-dependent limit to cell divisions prevents early cancer clones from progressing into frank cancers and constitutes a powerful tumor suppressor pathway. Now, a new study in Molecular Cell demonstrates that replicative senescence depends entirely upon the ATM kinase, a signaling protein that responds to DNA breaks and is crucial for maintaining genomic stability.
The new findings also address the old question of why cells stop dividing sooner under the high oxygen conditions used in laboratories, compared to when they are cultured under the low oxygen conditions of the human body. When oxygen levels are high, the researchers found, ATM becomes hyperactive, responding vigorously to DNA breaks and reducing the cell's tolerance for short telomeres.
"Our results have illuminated the mechanism underlying the aging of human cells through replicative senescence," says Titia de Lange, head of the Laboratory of Cell Biology and Genetics. "These insights are critical for understanding how this tumor suppression pathway prevents cancer."
Creating the right conditions
By halting the growth of cells with compromised telomeres, replicative senescence, prevents early-stage cancers from continuing to divide. The arrest is triggered when telomeres become so short that they can no longer recruit sufficient TRF2, a shelterin protein that keeps telomeres protected. When there is insufficient TRF2, telomeres resemble a DNA break, activating the cascade of events that grind cell division to a halt.
"Replicative senescence is a remarkably effective tumor suppressor pathway," de Lange says. "We know this from patients with long telomeres in which this system does not work properly. These patients can get as many as five different cancers before the age of 70, indicating that in people with normal length telomeres, the telomere tumor suppressor pathway prevents many cancers."
Despite the importance of this cancer prevention program, a number of long-standing questions about replicative senescence remained unanswered. It wasn't clear which DNA damage signaling pathway actually mediated the cell division arrest-studies hinted that both ATM and ATR might be involved-and an even bigger questions surrounded the role of oxygen. For decades, researchers observed that cells grown in standard lab conditions, at about 20 percent oxygen, hit senescence far sooner than cells grown at the much lower oxygen levels found in the body, somewhere between 1 and 8 percent. Yet the leading explanation, that high oxygen accelerates telomere erosion, had been ruled out, leaving no satisfying answer as to how oxygen levels modulated cellular aging.
To understand why high oxygen levels accelerate cell aging, de Lange and colleagues tracked senescence in primary human fibroblasts grown at 3 percent or 20 percent oxygen. Growing cells under low oxygen conditions was no easy task. Working at 3 percent oxygen meant that even ordinary tasks-moving plates, lysing cells, or adding reagents-had to be done fast and with care so that the cells would not be exposed to atmospheric oxygen.
"Any time the cells or the reagents are outside of the special low oxygen incubator, they are exposed to 20 percent oxygen which can change the molecular environment within minutes," says Alexander Stuart, a former graduate student in the de Lange lab who now holds a postdoc position at Harvard. "That means you're often in a race to do all the standard protocol steps extremely quickly so you can keep the samples at low oxygen as much as possible."
Stuart first determined that ATM alone enforces senescence both at 3 percent and 20 percent oxygen. Inhibiting ATM (or overexpressing TRF2) allowed cells to grow beyond their usual expiration date, and blocking ATM signaling in arrested cells brought them back to life, demonstrating that the arrest is reversible and entirely ATM-dependent.
The impact of high oxygen
Stuart and de Lange then discovered that high oxygen accelerates cellular aging because it creates a hyperactive form of ATM. Further work revealed that the lifespan gap between cells grown at low and high oxygen reflects how well cells can tolerate very short telomeres. At 3 percent oxygen, cells can keep dividing even after many of their telomeres become extremely short; they only stop when moved to 20 percent oxygen, where ATM becomes so reactive that it treats those short telomeres as urgent DNA damage and forces the cells into senescence.
"I don't think of it as low oxygen extending the lifespan of human cells-that's the physiological state of our bodies. Rather, the question was: why do high oxygen conditions shorten cellular lifespan? One could then extend that question: why aren't high oxygen conditions accurate systems for studying senescence?" Stuart says. "We've now shown that high oxygen represents a hyperactive ATM setting, which leads to fewer divisions than cells would naturally undergo."
Stuart and de Lange traced the effect of oxygen on ATM to molecules known as reactive oxygen species (ROS), which, counterintuitively, are more prominent under low oxygen conditions. ROS caused ATM molecules to lock together through chemical bridges called disulfide bonds, creating dimers that can't respond to DNA breaks or withered telomeres. With the help of Ekaterina V. Vinogradova, head of Rockefeller's Laboratory of Chemical Immunology and Proteomics, they identified where in ATM these disulfide bonds are formed and showed that one of those bonds is needed for the regulation of ATM by oxygen.
Together, the findings confirm that the replicative senescence pathway is controlled entirely by ATM, and that ATM behaves very differently under high oxygen conditions. The results may have immediate implications for scientists studying the DNA damage response in the lab. "Studying that in human cells cultured at 20 percent oxygen means you're basically studying the ATM kinase under hyperactive conditions," de Lange says. "We're not saying that everybody should switch to working at low oxygen, because it's very hard to do, but it may be a good idea to verify that what is observed at 20 percent also holds at 3 percent oxygen."
In the meantime, de Lange's findings also have implications for human disease. Most tumors experience oxygen levels that suppress ATM activity, enabling cancer cells to tolerate exceptionally short telomeres that would otherwise be unendurable. Therapies focused on restoring ATM function in these settings could potentially force vulnerable malignant cells into growth arrest.
"Telomere shortening represents a very important cancer prevention program." de Lange says. "Questions about how this system works have been at the heart of the work in my lab for years now, and we'll continue to dig deeper into this pathway."