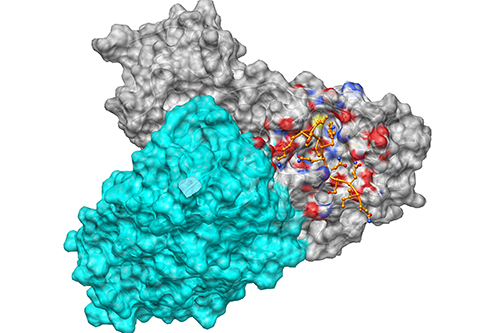
Mpro dimer from SARS-CoV-2 in complex with the inhibitory peptide (13) following 100 ns of molecular dynamics simulation University of Bristol
A new advance towards the development of drugs specifically designed to inhibit a key SARS-CoV-2 enzyme is reported in the Royal Society of Chemistry's leading journal, Chemical Science. The international team, led by scientists from the Universities of Oxford and Bristol, has designed new peptide molecules and shown that they block (inhibit) the virus's main protease [Mpro] - a prominent SARS-CoV-2 drug target.
Once SARS-CoV-2 invades a healthy human cell, the virus's own genetic material commandeers the infected cell's machinery, forcing it to make new copies of the virus. A vital step in this viral life cycle involves cutting a very long 'polyprotein' into its constituent viral proteins. SARS-CoV-2 has two molecular machines called protease enzymes that act as 'molecular scissors'. One of these, called the main protease, or 'Mpro' for short, has the vital role of chopping up the polyprotein, cutting it at 11 different places.
In the early days of the pandemic lockdown, Professor Garrett Morris at the University of Oxford, brought together a group of scientists to try to understand Mpro, with the aim of helping develop drugs against COVID-19. Meeting weekly over many months by Zoom, this group combined their computational and experimental expertise, and grew to include scientists from several different countries. From Bristol, this included Professors Adrian Mulholland and Jim Spencer, Dr Deborah Shoemark, PhD student Becca Walters, and other colleagues. Using a wide array of computational molecular modelling techniques including interactive molecular dynamics in virtual reality, quantum mechanics, peptide design and protein-ligand interaction analysis, the scientists were able to build an atomic level picture of the structure, dynamics and interactions of Mpro.
From these models, the team were able to find how the viral Mpro 'molecular scissors' work. They then designed new peptides, which are short pieces of protein, as inhibitors, to bind tightly to Mpro and prevent it from working, stopping the virus dead in its tracks. But did they work?
All 11 protein cut sites and four of these designed peptides were synthesized and tested in the Chemistry Research Laboratory at the University of Oxford. Experiments, led by Professor Chris Schofield at Oxford, showed that the novel peptides - designed by Dr Deborah Shoemark, with software developed in Bristol - not only bound to the molecular scissors, but they outcompeted the natural protein cut sites and so inhibited Mpro.
Adrian Mulholland, Professor of Chemistry at the University of Bristol and one of the study's lead authors, said: "Despite the development of successful vaccines in record time, new antiviral drugs are desperately needed. To date there are no drugs designed specifically to target COVID-19. Computational molecular modelling can really help with this. As we've shown here, computational design can produce molecules that actually stop the Mpro enzyme from working."
Dr Deborah Shoemark, Senior Research Associate (Biomolecular Modelling) in the School of Biochemistry, added: "It has been great to work together on this, combining our ideas and methods to get a really detailed picture of how this viral enzyme works – and to design molecules that actually stop it from working. Understanding Mpro specificity provides the potential to exploit vulnerabilities of the SARS-CoV-2 virus that may provide routes to new antivirals."
Professor Mulholland added: "This collaboration has really shown how sharing of models, data and expertise can help get understanding and make progress much more quickly. Garrett (Morris) built a fantastic team, and it has been exciting to work together on this. It's how science should be done – particularly in the face of pressing problems like the COVID-19 pandemic."
The study was funded through several grants including support from the EPSRC, BBSRC and the Wellcome Trust.
Paper
'Discovery of SARS-CoV-2 Mpro peptide inhibitors from modelling substrate and ligand binding' by Chan et al preprint on bioRxiv and in Chemical Science





