Long-term hematopoietic stem cells (LT-HSCs) generate and maintain lifelong blood cell production through their self-renewal and multi-lineage capacity. In the steady state, this rare population of adult stem cells is quiescent, resulting in decreased reactive oxygen species (ROS) and thereby preserving genome integrity. Driving the quiescent HSCs into cycling due to physiological stress such as infection or chronic blood loss leads to a rapid increase in the levels of oxidative DNA damage, including apurinic / apyrimidinic (AP or abasic) sites. Unrepaired AP sites in the replication-associated single-stranded DNA (ssDNA), where no complementary strand is available, can lead to DNA double-strand breaks (DSBs), a lethal DNA lesion, upon their removal by the AP endonuclease. They may also be processed by translesion synthesis (TLS) polymerases leading to mutations. Therefore, repairing or shielding the burst of AP sites in HSCs in a timely manner is essential for protecting their genome integrity and regenerative capacity. However, this mechanism remains obscure.
Prof. SHEN Li from the Zhejiang University Life Sciences Institute conducted collaborative research with JU Zhenyu from Jinan University and WANG Hu from Hangzhou Normal University in this regard. Their findings appeared in an open-access article entitled "HMCES safeguards genome integrity and long-term self-renewal of hematopoietic stem cells during stress responses" in the journal Leukemia on January 17.
HMCES (5-Hydroxymethylcytosine Binding, ES Cell Specific) has been identified as a sensor and shield of AP sites in ssDNA. It can covalently cross-link to AP sites in ssDNA via its conserved SOS response associated peptidase (SRAP) domain. This action results in the formation of DNA-protein crosslink intermediates and effectively protects genome stability by shielding AP sites from TLS polymerases and the AP endonuclease, which would otherwise generate DSBs or mutations.
In the study, researchers generated Hmces-knockout mice and examined the function of HMCES in hematopoiesis. Although HMCES was highly expressed in the bone marrow, Hmces-deficient mice exhibited normal development and steady-state hematopoiesis, loss of HMCES induced impaired long-term self-renewal capacity and increased DNA damage in LT-HSCs during stress hematopoiesis when ROS levels were on the rapid rise. Besides increased sensitivity to activation-induced intrinsic DNA lesions, researchers also found that sublethal irradiation led to more severe consequences in Hmces-deficient mice than in wildtype mice, including bone marrow failure and death. Moreover, Hmces-deficient LT-HSCs also exhibited mild but broad attenuation of DDR and repair pathways, including the fanconi anemia (FA) pathway. As a result, HMCES safeguarded AP sites on ssDNA and regulated DNA repair pathways, thereby ensuring that LT-HSCs could maintain their genome integrity and self-renewal capacity during the process of stress hematopoiesis.
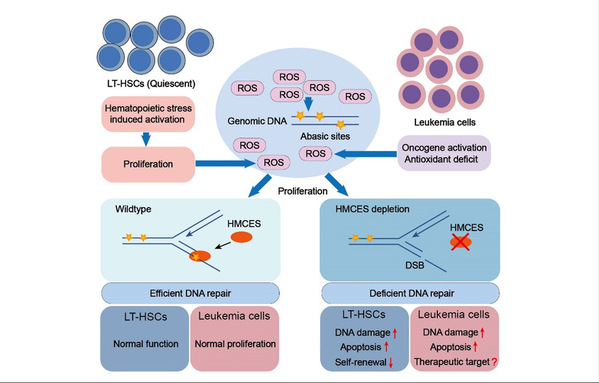
HMCES were also shown to protect leukemia cells from oxidative DNA damage and were thus essential for leukemia cell survival. The elevated expression of HMCES occurred frequently in acute lymphocytic leukemia (ALL) and was associated with poor prognosis. Given that HMCES is not required for normal hematopoiesis in the steady state, it may serve as a potential selective target against ALL while sparing normal hematopoiesis.
"The study not only reveals a critical role of HMCES in protecting the genome integrity and long-term self-renewal of HSCs during stress responses, but it also highlights its clinical significance in ALL and positions HMCES as a potential therapeutic target against ALL," said Prof. Shen.





