Body cells burn off fat reserves when nutrient supply from food ceases.
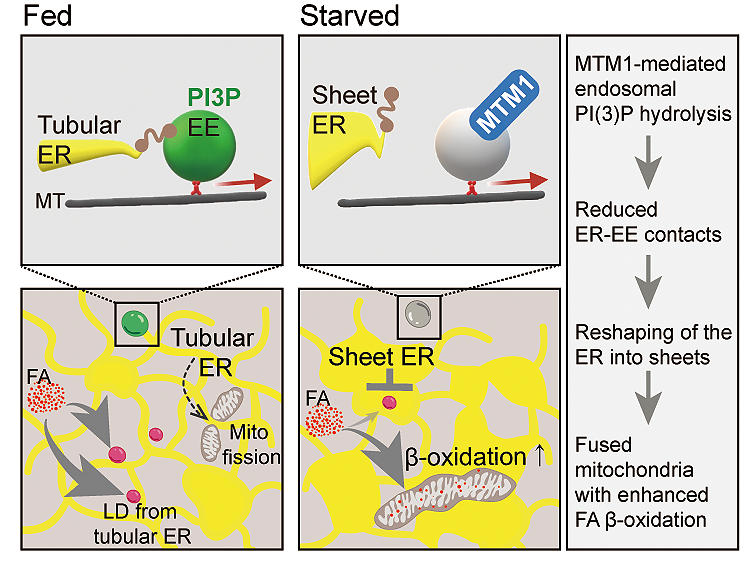
Role of MTM1-mediated endosomal PI(3)P signaling in mitochondrial metabolic rewiring via reshaping the ER in response to starvation. In fed cells, early endosomes form contacts with ER tubules. Tubular ER membranes facilitate mitochondrial fission and serve as source for lipid droplet formation. Nutrient starvation-induced hydrolysis of endosomal PI(3)P by MTM1 reduces membrane contacts between the tubular ER and early endosomes. The resulting loss of peripheral ER tubules induces mitochondrial network formation and the delivery of fatty acids to mitochondria to sustain cellular energy supply. | Visualization: Wonyul Jang
A team led by Professor Volker Haucke and Dr. Wonyul Jang from the Leibniz-Forschungsinstitut für Molekulare Pharmakologie (FMP) has now discovered a previously unknown mechanism for how this "starvation response" is triggered, and what can inhibit it. The results have been published in the renowned international journal Science.
In order for the body to function, cells need a constant supply of energy. During phases of starvation, when no nutrients are taken up from food, the cellular metabolism must adapt to ensure a sustained supply of energy.
Researchers from the FMP have gained new insights into this fundamental mechanism in human cells while investigating a rare genetic muscular disorder - X-linked centronuclear myopathy (XLCNM). This disease, which usually affects boys, involves a defective gene on the X chromosome, resulting in a developmental disorder of the skeletal muscles. This muscle weakness is so severe that, in many cases, affected children require ventilatory support and are wheelchair-bound. Affected individuals do not survive beyond the age of 10 to 12 years; in severe cases, they die soon after birth.
The genetic defect present in this disease affects the lipid phosphatase MTM1. This enzyme controls the turnover of a signaling lipid on endosomes, vesicle-like structures in cells involved in the sorting of nutrient receptors. It was during the study of the structure of mutant human muscle cells from patients that the researchers discovered changes in the endoplasmic reticulum (ER), a membrane network that spans the entire cell. In healthy cells, the ER forms a large interconnected network of "flattened" membrane-enclosed sacs near the nucleus of the cell and narrow tubules in the cell periphery. In diseased cells, tubules and, moreover, the membrane-enclosed sacs appear perforated. The researchers found a very similar accumulation of narrow ER tubules and perforated membrane-enclosed sacs in starved cells, in which MTM1 was genetically inactivated.
"Muscles are highly sensitive to starvation; their energy reserves are soon depleted. We therefore began to suspect that the defect in cells from XLCNM patients might be related to an incorrect response to starvation," reported Volker Haucke. When cells are starved, amino acid deficiency occurs. As a result, the researchers found, the ER undergoes changes in shape in healthy cells - the outer narrow tubules regress and are converted into flat membrane-enclosed sacs. This altered structure of the ER enables the mitochondria - spherical organelles that supply the cell with energy (adenosine triphosphate, ATP) and are in contact with the ER - to fuse together. "Such greatly enlarged 'giant mitochondria' are much better able to metabolize fats," explained Dr. Wonyul Jang, lead author of the study.
However, fats cannot be transported or burned efficiently in cells deficient in MTM1. The endosome controlled by MTM1 plays the key role in this process. In healthy cells, starvation reduces the contact points between endosomes and the ER, allowing the latter to reshape as a result. In cells of XLCNM patients, however, no contact site reduction occurs: the endosome exerts a "pulling force" on the ER, causing the stabilization of peripheral tubules and fenestration of the membrane-enclosed sacs. Since peripheral ER tubules are responsible for mitochondrial fission, mitochondria remain small in the absence of MTM1. In this shape, they are much less able to burn storage fats, resulting in severe energy deficiency in the cell (1).
"We have found a completely new mechanism for how different compartments in the cell communicate with each other such that cell metabolism adapts in response to food supply," summarized Volker Haucke. In light of this, the current study shows that starvation is totally harmful to the muscle cells of XLCNM patients. They need constant food intake to prevent muscle proteins from being broken down into amino acids. FMP researchers were able to show in a second study (2) that defects due to a loss of the lipid phosphatase MTM1 can essentially be repaired by inactivating the "opposing" enzyme, the lipid kinase PI3KC2B. Only time will tell if this will work in XLCNM patients. The team led by Volker Haucke is currently working to find a suitable inhibitor that can suppress PI3KC2B activity. They have already demonstrated in cell culture that this is possible in principle.
(1) Jang, W., Puchkov, D., Samso, P., Liang, Y.T., Nadler-Holly, M., Sigrist, S.J., Kintscher, U., Liu, F., Mamchaoui, K., Mouly, V., Haucke, V. (2022)
Endosomal lipid signalling reshapes the endoplasmic reticulum to control mitochondrial function. Science [advance online]
(2) Samso, P.*, Koch, P.A.*, Posor, Y., Lo, W.T., Belabed, H., Nazare, M., Laporte, J., Haucke, V. (2022)
Antagonistic control of active surface integrins by myotubularin and phosphatidylinositol 3-kinase C2b in a myotubular myopathy model.
Proc Natl Acad Sci USA 119, e2202236119





