With obesity rates projected to continue rising globally, there is an urgent need to understand the intricate mechanisms that contribute to metabolic diseases such as diabetes. Addressing this critical issue head-on, Jiyoung Park and her research team in the Department of Life Sciences at UNIST has led a groundbreaking study on endotrophin-an extracellular matrix protein that plays a pivotal role in adipose tissue homeostasis.
The research team's findings shed light on how intracellular endotrophin levels impact autophagy-a natural cellular process responsible for removing unnecessary or dysfunctional components. In obese individuals, elevated levels of endotrophin drive detrimental effects within fat cells by disrupting autophagic flux-leading to inflammation and insulin resistance associated with diabetes.
"Understanding the relationship between obesity-related metabolic dysfunction and endotrophin presents significant implications for developing targeted treatments for metabolic diseases," emphasized Professor Park Ji-young.
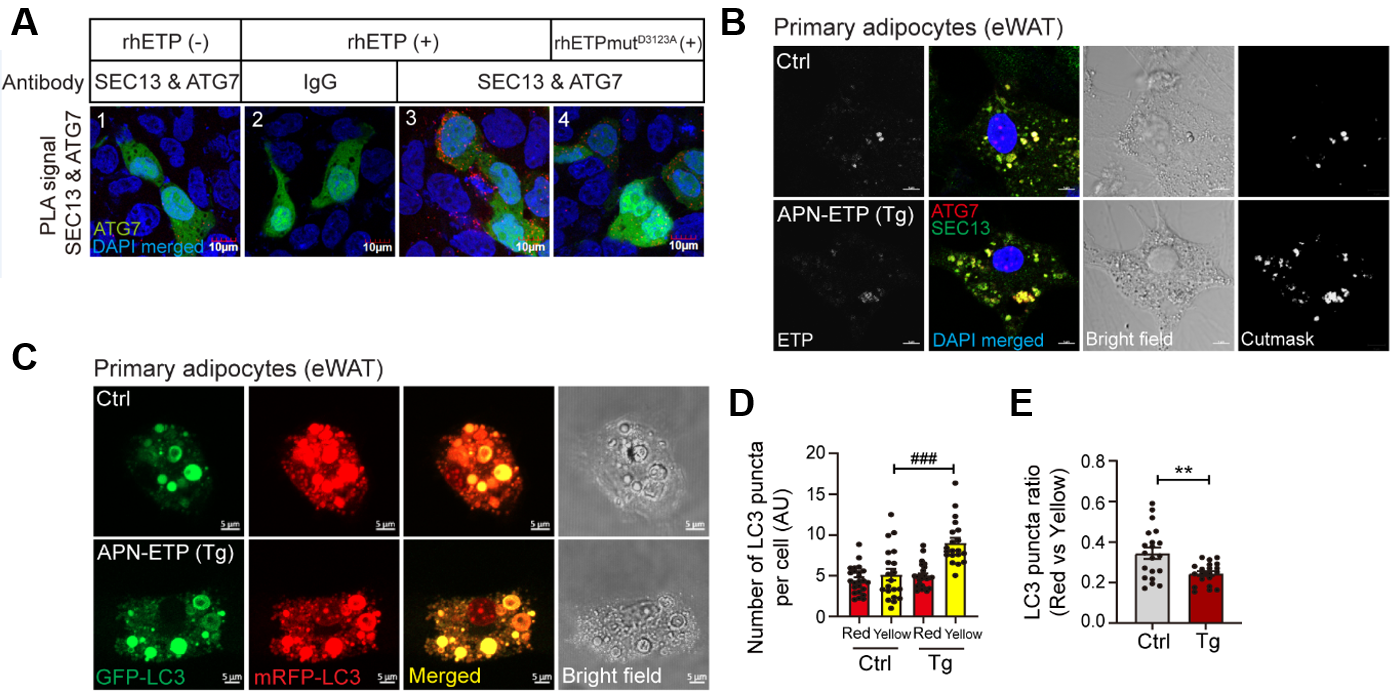
Figure 1. Endotrophin-mediated interaction between SEC13+ COPII and ATG7 contributes to increasing autophagosome formation and impairs autophagic flux in adipocyte in obesity. PLA utilizes the specificity of a secondary antibody conjugated with the PLA probe, which amplifies the interacting protein complex shown by red puncta. GFP+ area indicates the cytoplasmic region.
Endotrophin was first discovered by Professor Park in 2012, as a key player responding to metabolic stress in obese individuals. The recent study delved deeper into understanding how endotrophin enters and influences fat cells under both lean and obese conditions through meticulous comparative analysis.
In cases of obesity, it was observed that endocytosis-the process through which extracellular substances are internalized into cells-facilitates the accumulation of endotrophin within fat cells. This accumulation not only promotes the formation of autophagosomes involved in autophagy but also disrupts their degradation process-ultimately leading to cell death, inflammation, and insulin resistance.
Furthermore, the research team uncovered specific interactions between proteins involved in protein transport (SEC13) and autophagy (ATG7), which play crucial roles in driving excessive autophagosome formation triggered by endotrophin within fat cells. This dysregulation of autophagy further exacerbates the adverse effects on adipocyte health.
Excitingly, the study also demonstrated that inhibiting ATG7 protein function or neutralizing endotrophin using the siRNA system-specifically designed to interfere with gene expression-significantly improved metabolic diseases associated with obesity. These findings pave the way for potential future treatments targeting endotrophin-mediated autophagic flux impairment in obesity-related conditions. "The accumulation of endotrophin in cells can serve as a biomarker indicating an imbalance in extracellular matrix homeostasis," remarked Professor Park.
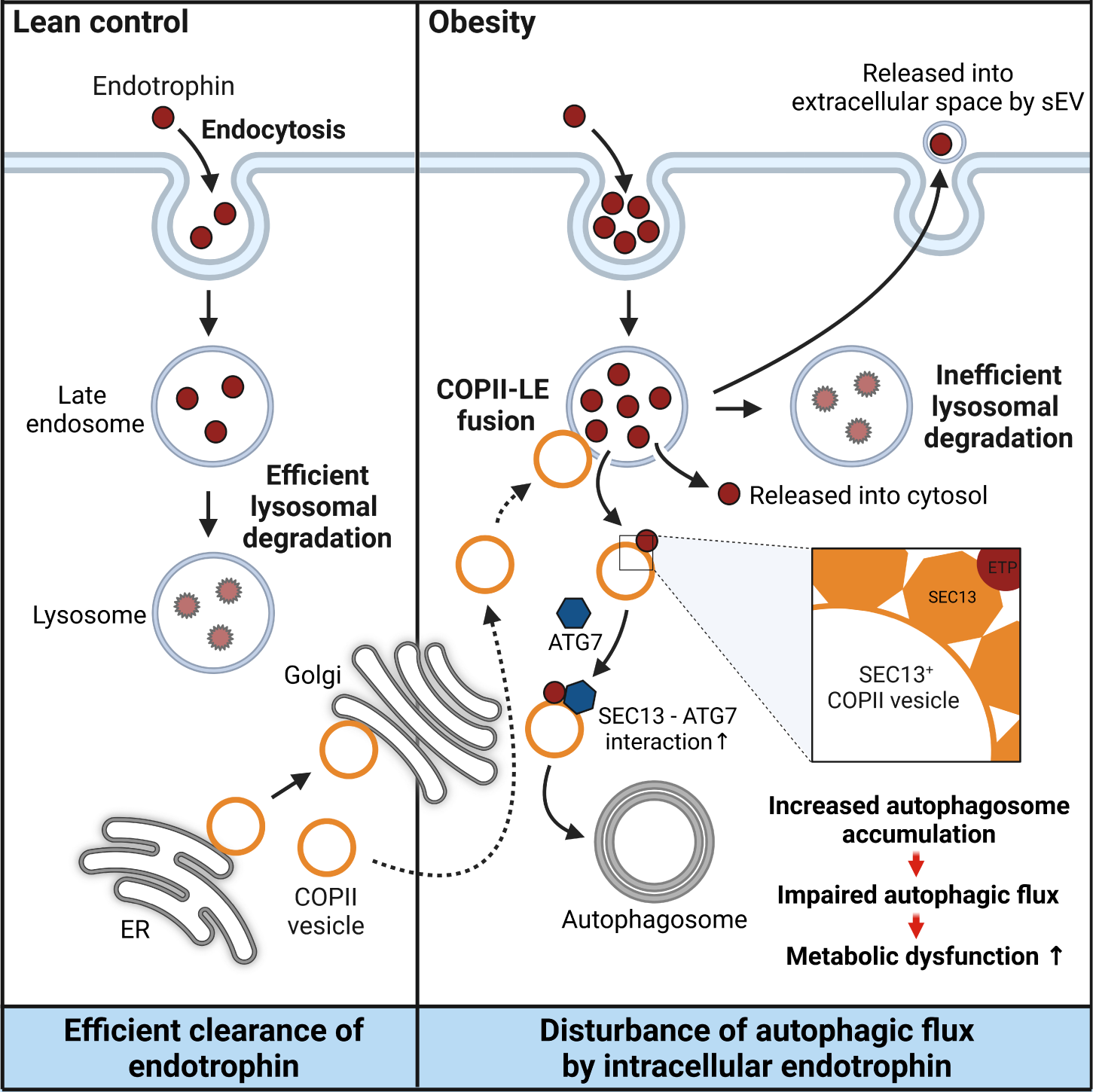
Figure 2. This is a single, concise, pictorial and visual summary of the main findings of the article.
The study findings have been published ahead of their official publication in the online version of Metabolism: Clinical and Experimental-a prestigious international academic journal specializing in endocrine metabolism-on June 10, 2023. This work has received support from the National Research Foundation (NRF) of Korea, funded by the Ministry of Science and ICT (MSIT).
Journal Reference
Jiyoung Oh, Chanho Park, Sahee Kim, et al., "High levels of intracellular endotrophin in adipocytes mediate COPII vesicle supplies to autophagosome to impair autophagic flux and contribute to systemic insulin resistance in obesity," Metabolism (2023).





