Drug discovery is like molecular Tetris. Chemists snap atoms together, adjusting the pieces until everything fits and suddenly, a molecule makes a promising new medicine. Normally, creating better molecules consumes huge amounts of time and money.
In a new study, researchers used machine learning to build a smarter prediction system that could speed up the process at a fraction of the cost.
"Sometimes we use sophisticated, physics-based computational chemistry tools to understand novel reactions. However, these tools are too expensive to make predictions on thousands of potential new molecules," said Simone Gallarati, the study's co-lead author and joint postdoctoral researcher at the University of Utah and the University of California, Los Angeles. "We wanted to train statistical models that were 'smart' enough to make accurate predictions on untested reactions, but also as cheap as possible."
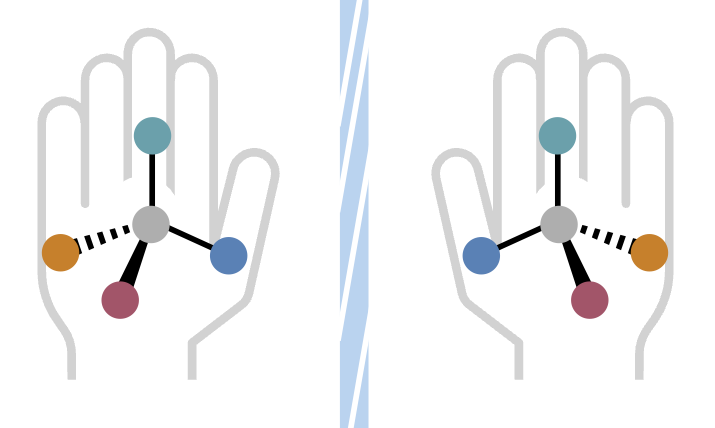
Molecules can exist as mirror images, a property known as "handedness." Left- versus right-handed forms are crucial; one may heal, the other might harm. Chemists need to find just the right set of tools-catalysts, ligands and substrates-to ensure they construct the correct version.
The new system acts as a high-tech filter that can screen tens of thousands of chemical structures to predict how the pieces will come together to produce one "hand" of a molecule over another. The workflow provides a cost-effective way to convert the reaction's components into numerical data a computer can analyze, building the framework for machine-learning predictions.
With surprisingly little input the model reliably forecasted how the components would behave, cutting down the time, energy, and expense spent testing reactions in the lab.
"Most AI requires enormous amounts of data to train models on. That's a problem in chemistry by which obtaining high-quality, large datasets from experimental work is very expensive and extremely time consuming," said Matthew Sigman, chemist at the U and coauthor of the study. "The coolest thing about this tool is that it allows someone to collect smaller bits of data, build reasonably good models and make accurate predictions for known reactions, and also transfer predictions to reactions that the models haven't seen yet."
The study was published as an accelerated preview in the journal Nature on Feb. 11, 2026.
High-tech filter

The researchers centered the workflow around asymmetric cross-coupling reactions, a powerful toolkit for drug development. The reactions stitch together two carbon-based molecular fragments, using a metal catalyst to build more complex compounds. The reactions are called asymmetric because they're designed to favor one "handed" version of the molecule. Chemists often produce both versions but without guidance, the experiments will yield a 50/50 split. In contrast, asymmetric reactions deliver, say, 95% of the desired form and just 5% of the unwanted mirror image.
Asymmetric cross-coupling reactions generally require at least three elements-a metal, a ligand and substrates. The metal catalyst does the heavy lifting by joining carbon-based molecules to build the product. A ligand binds to the metal, controlling which side of the molecule reacts, influencing the three-dimensional orientation of the product. The ligand is arguably the most important element to control a molecule's handedness.
To train their model, Gallarati and the team identified four academic papers on asymmetric reactions-coauthor Abigail Doyle's and Sigman's past work included-that all used nickel-based catalysts with different ligands. Those results were the only training data for the workflow. Then, the team asked the system to predict the outcomes of hypothetical components not included in the training data. They added a series of increasingly challenging tasks that forced the algorithm to make predictions with materials that were increasingly dissimilar to the original training data. The team tested the prediction in the Doyle lab, an effort led by Erin Bucci, the study's co-lead author and doctoral student at UCLA.

"As a lab-based chemist, this tool is extremely valuable for saving time spent running experiments," Bucci said. "For example, instead of running 50-60 reactions, we are now able to run 5-10, potentially saving weeks or months. Each reaction component we test in the lab needs to either be purchased or made from scratch-this tool greatly cuts the amount of money I would typically spend on materials."
While the authors tested the tool in the context of new nickel-based reactions, the workflow can apply across fields and even deepen our understanding of chemistry itself.
"One of the nice things about the workflow is-it's not a black box," said Abigail Doyle, chemist at UCLA and coauthor of the study. "We can learn something about the chemistry from the predictions, even if they're off. We apply our chemistry expertise to help learn something we wouldn't have learned without the tool."
The pharmaceutical industry would immediately benefit from a tool like this, Sigman added. Say a company needs to deliver large quantities of a compound for a clinical trial and they want to apply a reaction already in the literature. But it's never been done on their specific compound target.
"This is where this tool could be highly applicable," he said. "Optimizing a reaction and the time-cost is the value proposition when you build a drug. This streamlined process could make the difference when they need to take a molecule from phase one to phase two."





