
Arthur, Arturito, and Olga Estopiñan. Photo courtesy of the Estopiñan family.
When Arthur Estopiñan first spoke with Columbia neurologist Michio Hirano about his 18-month-old son's rare genetic disease, he couldn't believe his ears. It was 2012, and Estopiñan had been calling doctors around the country, trying to find a treatment to save his son, Arturito, who had been diagnosed months earlier with a rare and usually fatal disorder.
Arturito's disorder, caused by a TK2 enzyme deficiency, rapidly weakens and destroys muscles. All the other doctors Estopiñan called said no treatment existed and recommended that Estopiñan and his wife Olga take their son home, make him as comfortable as possible, and wait for the inevitable end.
"Dr. Hirano said he had an experimental treatment, and I nearly fell out of my chair. I repeated the diagnosis to make sure he had understood," Estopiñan recalls. "He said, 'I understood what you said. And yes, we have a clinical path forward.'"
Hirano was also a bit surprised to have a potential treatment in hand. Just a few years earlier, Hirano had created a line of mice with the disease, a step essential to identifying therapeutic possibilities.
"When you try to replicate a human disease in mice, it's hit or miss," Hirano says. "It was fortunate for us that in our first attempt our mice had a very similar disease, and we could begin characterizing the disease and testing potential treatments."
With the mice, Hirano could see that the TK2 deficiency robbed muscle cells of building blocks needed to maintain their mitochondrial DNA. He also found that administering these building blocks could potentially restore mitochondrial health and prevent degeneration of muscle cells.
"We started using therapies in the mouse model, and they worked. Depending on the dose we used, it doubled or tripled the lifespan of the mice," Hirano says. The compounds were supplied by chemical manufacturing companies, and within a couple years, Hirano's collaborators were providing chemical-grade material to patients in Spain.
"A second chance at life"
Arturito arrived in Hirano's clinic just as the first patients in Spain started the experimental therapy . "When Arturito came to us, he was in terrible condition, unfortunately, and in the terminal stage of the disease. He couldn't lift his arms or legs, he couldn't lift his head; he was like a rag doll," Hirano recalls.
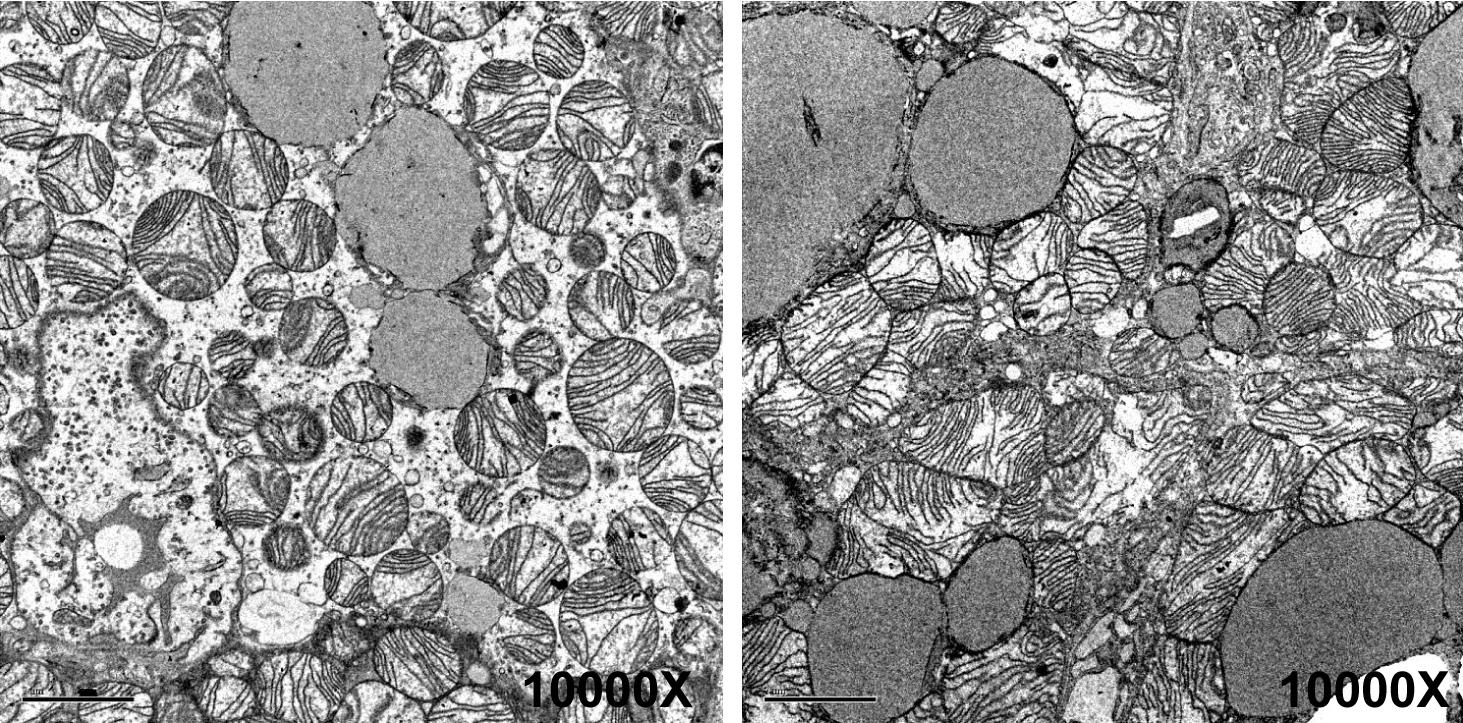
In TK2 deficiency, movement is lost as mitochondria inside cells become dysfunctional. The left image shows normal mitochondria in an unaffected mouse; the right image shows abnormal mitochondria from a mouse with TK2 deficiency. Images from Villarroya, et al. PLoS ONE 6(12): e29691.
It was too soon to tell if the therapy was helping the Spanish patients. If the FDA granted approval for emergency compassionate use, Arturito would be the first person in the U.S. to try the therapy. "Arturito was deteriorating fast, and we were witnessing this every day," Estopiñan says. "We knew the drug had only been given to mice and two patients in Spain. Dr. Hirano gave us hope, but he was very realistic; he didn't know if it would work in Arturito."
When he came home from the hospital after receiving the first doses, Arturito could only move his eyes. "We started to see improvements, but it was extremely slow," Estopiñan says. The pace picked up when Hirano increased the dose and then switched to the therapy's second generation. By age 5, he could move his hands and feet. At 10, he was sitting on a tricycle.
Now 15, Arturito attends school, where he steers his own motorized wheelchair down the hallways to reach his classes and always makes the honor roll. Like most teenagers, he enjoys creating videos on his iPad and solving his parents' computer and tech problems. And he continues to get stronger with intensive physical therapy; now he is working to strengthen his leg muscles so that he can walk with a walker.
"Obviously, without this treatment, my son would not be here today," Estopiñan said in a recent Muscular Dystrophy Association video. "Dr. Hirano and his team gave my son a second chance at life."
Helping others
Over the years, the Estopiñans have helped raise money for Hirano to continue his research and buy compounds for the treatment of other patients.
"We were able to extend compassionate use treatment to so many other TK2D patients and continue refining the treatment," Hirano says. "Their support helped us to eventually get funding from the Muscular Dystrophy Association and then NIH to continue the progress."
By 2019, Hirano had treated about 20 patients and then opened a phase 2 clinical trial, a necessary step to obtaining FDA approval. A small biotech company (now owned by UCB in Belgium) produced the compounds and organized the trial.

Michio Hirano
Based on the results of that trial, the FDA approved the therapy in November 2025 for TK2 deficiency patients whose symptoms emerge at or before age 12.
"More than 100 patients are now being treated, and we've seen some dramatic responses, especially in young kids," Hirano says. "If patients are treated before their muscles break down, most regain motor functions, which is unusual in neuromuscular disease. Some kids who were unable to sit up before treatment are now walking and running.
The treatment also dramatically improves survival, a key factor considered by the FDA. Historically, patients with the earliest onset and most severe symptoms died by age 2. As of today, all participants in the trial but one are still alive.
Next steps
The success of the therapy, now called KYGEVVI, was made possible by dedicated families, doctors, scientists, funding agencies, and companies.
"This really took a whole village, from the post docs in the lab, to our clinical coordinators who helped with the expanded access and trial, to people from the companies who pushed it forward," Hirano says. "It's very rewarding for the whole team, and it feels great to develop something that really helps our patients."
It also took a bit of luck.
"We were fortunate at many steps: that we were able to produce a faithful mouse model, that there was a response to this therapy, and that the therapy not only worked in the mice, but in humans," Hirano says. "That doesn't always happen. A lot of therapies that look promising in the lab never make it to patients because they have toxic effects or just don't perform in people the way they did in animal models."
Hirano continues to search for even better treatments and has found that, in mice, a gene therapy combined with the drug therapy is markedly more effective. And he's testing a different therapy for another rare mitochondrial deficiency disease.
"Our success with KYGEVVI in TK2 deficiency is proof that we can reverse some of the changes caused by mitochondrial dysfunction. And that suggests we can succeed with other mitochondrial diseases," Hirano says. There are more than 400 different mitochondrial diseases currently known. Though each individually is rare, in aggregate, they affect about 1 in 5,000 people.
"Arturito, the Estopiñan family, and everyone involved in KYGEVVI are helping us open the door to improving the lives of many more patients," says Hirano.





