An experimental drug has been shown to alleviate symptoms of Huntington's disease and extend lifespan in mouse models. Further studies are required to determine whether these results may also apply to humans.
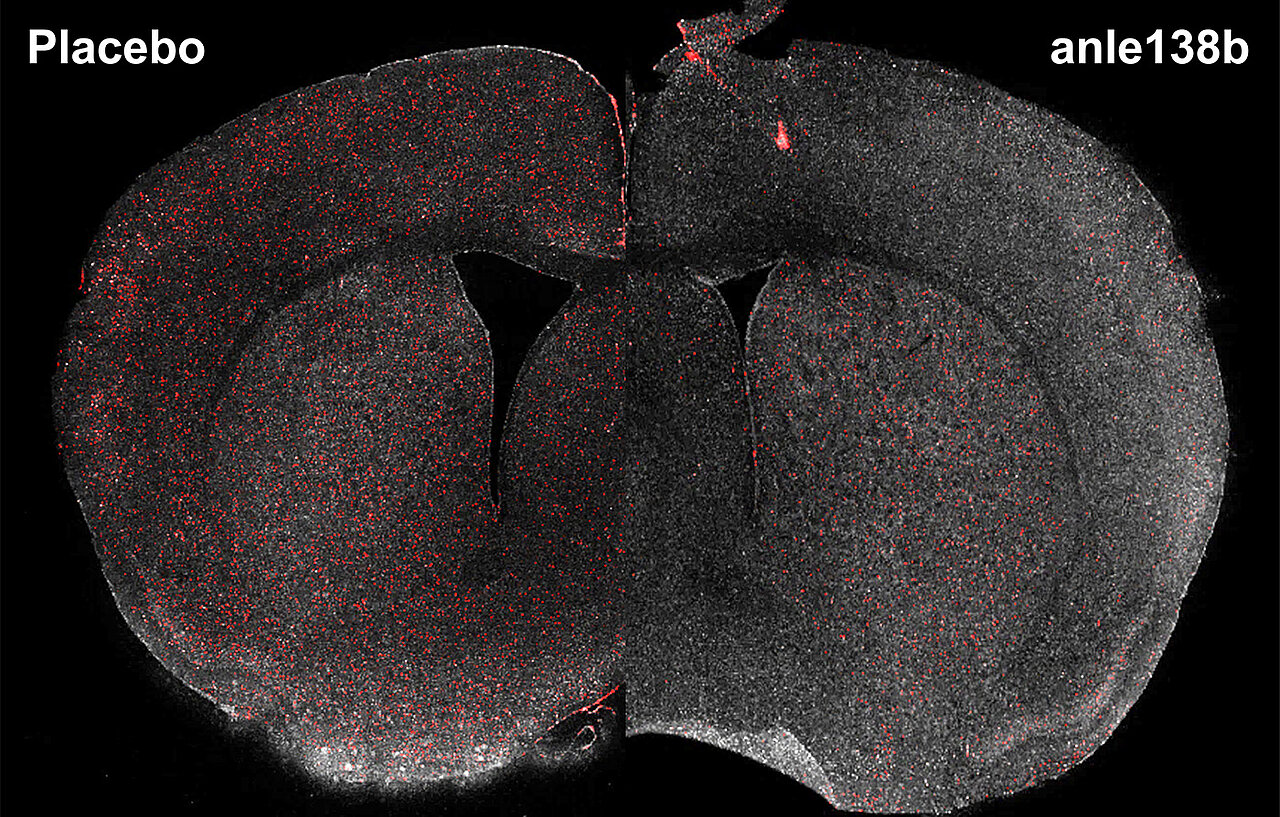
The hereditary disorder Huntington's disease has so far been considered incurable. Its clinical manifestations include impaired motor control and psychiatric symptoms. A new study offers promising insights. It shows that a specific drug candidate called anle138b can significantly reduce the toxic protein clumps in the brain that are characteristic of the disease.
Affected mice that were administered this compound retained their mobility for a longer time, their brains shrank less, and their lifespan was extended compared to untreated mice. Importantly, the compound not only alleviates symptoms but also addresses the underlying cause of the disease by preventing disease-specific harmful protein clumps from destroying nerve cells and their connections. These results were also confirmed in experiments with human stem cells from Huntington's patients.
Promising Therapeutic Candidate
These are the key findings of a study that has now been published in the journal EMBO Molecular Medicine. The study was led by Professor Irina Dudanova, who holds the Chair of Anatomy and Cell Biology I at the University of Würzburg since April 2026, and her doctoral student Miguel da Silva Padilha. The substance was developed by the teams of Christian Griesinger, Director at the Max Planck Institute of Multidisciplinary Sciences in Göttingen, and Armin Giese from the Ludwig-Maximilians University in Munich, now at MODAG GmbH. Other participants of the study come from the Max Planck Institute of Biological Intelligence in Martinsried and the University of Cologne.
"Our data show that specifically targeting toxic protein aggregates with the compound anle138b is a promising approach for stabilizing neuronal health in the long term," says Irina Dudanova, commenting on the study's findings.
Cellular waste destroys nerve cells
Background: Huntington's disease is an inherited movement disorder caused by a defect in a specific section of DNA, the gene that encodes the protein huntingtin. According to the health insurance organization AOK, approximately 10,000 people in Germany are affected by the disease. Several hundred new cases are diagnosed each year. A faulty repetition of the genetic code (known as CAG repeats) causes the huntingtin protein to take on an abnormal shape and form clumps.
The resulting protein aggregates can be thought of as a form of cellular waste that accumulates inside neurons. The protein aggregates disrupt vital cellular communication and lead to cell death, particularly in brain regions involved in for movement and cognition. An effective therapy that targets the underlying causes is not available. This is where the compound investigated anle138b comes into play, as it prevents the formation of the harmful structures.
The researchers investigated the efficacy of anle138b in two different mouse models: While one suffered from a severe, early-onset form of the disease, the other model mirrored the genetic situation in adult patients. The compound showed beneficial effects in both models.
A characteristic feature of Huntington's disease is the loss of the protein PDE10A, which is found almost exclusively in the nerve cells that die in this disease. The amount of PDE10A decreases dramatically long before patients show the first severe symptoms. "If PDE10A levels drop, that is a clear signal that the disease is progressing. The protein is therefore well-suited as a biomarker for Huntington's disease," explains Miguel da Silva Padilha. If less nerve cells die, then the PDE10A levels stay high. This is exactly what the scientists observed: as a result of anle138b treatment, the concentration of PDE10A remained high in both mouse models.
Efficacy Demonstrated in Human Stem Cells
A key milestone of the study is the confirmation of these effects in human cells. "In our experiments with induced pluripotent stem cells - that is, precursor cells derived from Huntington patients' cells - we also observed that the addition of anle138b reduced the amount of huntingtin aggregates," says Irina Dudanova.
Since the compound targets a fundamental mechanism of protein aggregation, it is also of interest for research on other neurodegenerative diseases. Corresponding studies in mouse models have been so promising that two years ago a large clinical trial was started for the treatment of multiple system atrophy - a Parkinson's-like disorder characterized by severe impairments of motor function, balance, and the autonomic nervous system.
Original publication
Anle138b ameliorates pathological phenotypes in mouse and cellular models of Huntington's disease. Miguel da Silva Padilha, Seda Koyuncu, Evangeline Chabanis, Sergey Ryazanov, Andrei Leonov, David Vilchez, Rüdiger Klein, Armin Giese, Christian Griesinger and Irina Dudanova. EMBO Molecular Medicine, DOI: 10.1038/s44321-026-00459-9





