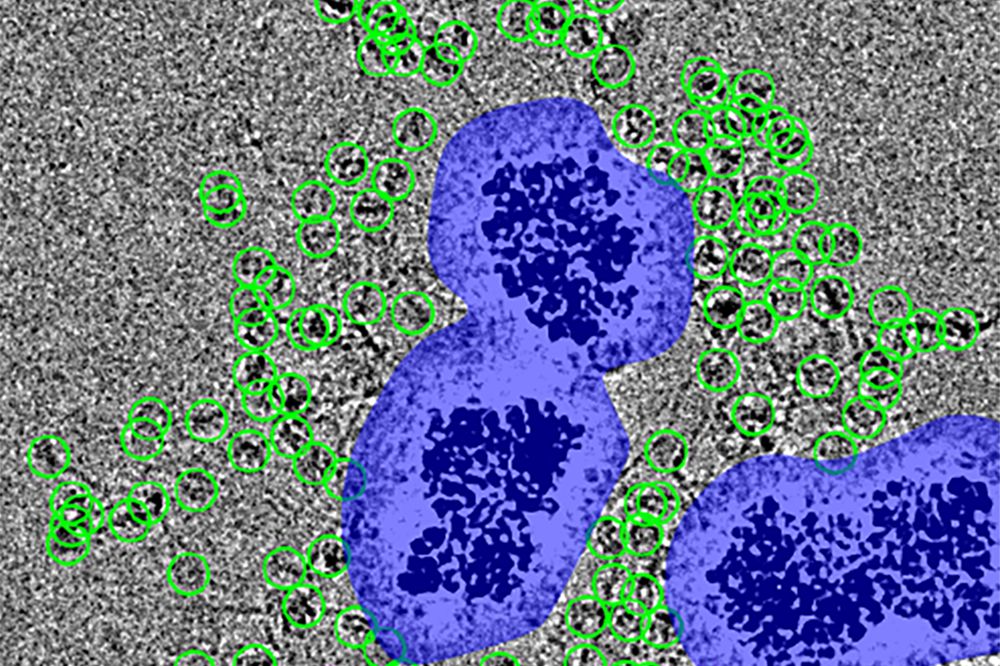
Particles (encircled in green) are spaced at a distance from the noise surrounding the nanobeads (purple), which would otherwise obscure them. (Courtesy of Funabiki Lab)
Sample loss has been a persistent problem in cryo-EM, a high-tech method for creating 3D models of molecules that reveals their inner structures. It occurs during an essential step of imaging preparation, when a sample is blotted with a filter to remove excess liquid. Frequently, much of the sample transfers to the filter, leaving little to nothing behind on the imaging platform for the electron camera to capture.
This sample loss has forced scientists to focus on protein complexes that are either naturally abundant or to artificially assemble them, which may lead to their being put together in a way that's different from their natural state.
Now researchers from The Rockefeller University have made dramatic improvements to the technology that greatly expand its uses. The method, called MagIC cryo-EM, uses magnetic beads to hold molecules in place, keeping the sample intact and reducing sample loss by a thousandfold. The researchers published their findings in eLife.
"Because MagIC cryo-EM requires only a small number of particles compared to the conventional method, we can use it to visualize a broader variety of molecules, such as proteins that are very rare, or hard to produce and purify," says first author Yasuhiro Arimura, a former research associate and currently a guest investigator in the Laboratory of Chromosome and Cell Biology, led by Hironori Funabiki. "It might be especially useful for the structural analysis of viruses and other infectious disease agents."
Revolutionary insights
When an innovative electron camera for cryo-EM led to a so-called "cryo-EM resolution big-bang" around 2016, it revolutionized structural biology. No longer did scientists have to spend years trying to find a condition in which the target proteins crystalize for X-ray structure analysis. Since then, cryo-EM has enabled scientists to visualize the interior workings of formerly impenetrable molecules for the first time, providing groundbreaking insights into some of the most fundamental mechanisms of life.
In cryo-EM, target particles are suspended in a liquid buffer that's dabbed onto a small gold disc that has a carbon film layer flecked with tiny holes. The disc is blotted to remove excess liquid and then plunged into liquid ethane, flash-freezing it. The result is a disc topped with a thin sheath of ice that's packed with molecules-at least when they aren't lost during the blotting process. An electron camera then takes millions of low-res 2D images of the molecules, and software assembles them into a high-res 3D structural model of the molecule.
But sample loss isn't cryo-EM's only shortcoming, says Arimura. It's also been inherently unsuitable for very small proteins. "The algorithm really struggles to find small proteins in these images, which have a low signal-to-noise ratio," he says. "Even if it can find them, it has a difficult time aligning them to form a composite 3D structure."
The scientists wanted to overcome cryo-EM's limitations precisely because they've repeatedly hit up against them in their own research. Funabiki's lab focuses on the molecular mechanisms of chromosomal inheritance, with a focus on the tightly compacted nucleosome. Thanks to its prototypical structure, the nucleosome can act as an excellent benchmark for cryo-EM technology development. At the same time, since the nucleosome is one of the major regulatory targets for genome function, it's important to understand how its structure is recognized and altered. Despite the large variations of genomic DNA sequences, most nucleosome structures have been analyzed using synthetic nucleosome-positioning sequences. Arimura wanted to capture the structure of nucleosomes and their regulators under native conditions.
Two problems, solved
The solution to sample loss turned out to be elegantly straightforward. Arimura coated tiny magnetic nanobeads with a corona of radiating "spacer" proteins that lure and capture target molecules; placed the beads inside the holes on the gold disc; and added a magnet to the bottom. This process tethered the target molecules to the magnetic beads, and the magnetic beads to the grid. With the particles securely in place, blotting could then be done safely, with far less sample loss.
"In traditional cryo-EM, the target molecules just float around in the buffer in a random distribution," says Funabiki. "But with our method, you can remove the extra liquid while the molecules remain in the buffer, ready for vitrification. It improves protein capture by a thousandfold."
At the same time, the spacers also compensate for any additional visual noise introduced by the nanobeads, Arimura adds. "If the particles were directly attached, they'd be lost in the white noise that comes from a high-power electron beam hitting the dense beads," he says.
As a result, these halos of proteins can now be imaged one by one for 3D model assemblage. They call their innovation MagIC-cryo-EM (Magnetic Isolation and Concentration).
Arimura also developed a method for addressing cryo-EM's second limitation-its inability to handle very small proteins. Called DuSTER (Duplicated Selection To Exclude Rubbish), this curation method excludes images of molecules with a low signal-to-noise ratio, allowing the algorithm to mold the 3D model from only higher quality images.
In the current study, the lab used DuSTER to image a molecule called NPM2 in its "open" form for the first time. A so-called chaperone protein, NPM2 and its target, the linker histone H1.8, are essential to the assemblage of nucleosomes, and "the H1 protein is really important for properly shaping the chromosome during cell division," Funabiki adds.
"Nobody knew how NPM2 binds to H1," Arimura says. "How do they recognize each other? There's been a lot of speculation and conflicting data in the scientific literature."
After DuSTER swept out the lowest-contrast images, more than 187,000 remained. These were assembled into the first-ever 3D model of NPM2 and H1.8. This structure, imaged in various formations, revealed distinct open and closed variants. H1.8, it turns out, binds to NPM2 during an open formation of the protein. Surprisingly, H1.8 also acts as a gatekeeper for other proteins' access to genomic DNA.
Future applications
These technological advances can be used for the structural analysis of a wide variety of protein complexes-including those of viruses, Funabiki says. To that end, the study was supported by the NIH and the SNF Institute for Global Infectious Disease Research at Rockefeller.
"Thes techniques will be very useful for the structural analysis of virus components, for example," Funabiki says. "These are notoriously difficult to generate a lot of, so putting painstaking effort into producing them only to have most of them become lost during blotting has been a significant issue."
Now an assistant professor and head of his own lab at the Fred Hutchinson Cancer Center, Arimura is further improving these approaches to expand their applications.
"We're already hearing from a lot of researchers around the world," he says. "We're very excited to see how our techniques may contribute to infectious disease research."





