Scientists at UCL have used computer-based imaging analysis to identify new patterns of lung damage in patients with Idiopathic pulmonary fibrosis (IPF), providing the first evidence of a previously unknown and life-limiting lung disease subtype.
IPF is a condition where scar tissue or fibrosis builds up in the lungs, making them thick and hard. It affects around 3 million people globally (around 32,500 people in the UK), with those affected typically aged between 50 and 70, and has a life expectancy of three to five years.
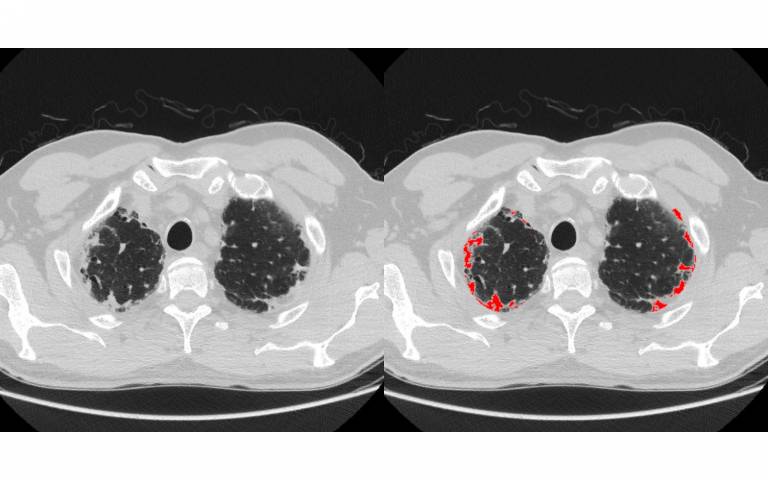
In this study, published in the Lancet's EClinicalMedicine, researchers at the Centre for Medical Image Computing (CMIC) at UCL, developed a computerised image-analysis algorithm, partially based on deep learning, to see if lungs affected by IPF may also have scarring patterns characteristic of another serious, though less well-documented, lung disease called pleuroparenchymal fibroelastosis (PPFE). It is not known for certain what causes either IPF or PPFE.
Explaining the research, lead author Dr Joe Jacob (UCL Division of Medicine) said: "Idiopathic pulmonary fibrosis is a disease that shrinks the lungs and is associated with a life expectancy comparable to many cancers.
"Doctors have suspected for some time that there are distinct subgroups or endotypes of IPF that have unique functional or pathobiological disease mechanisms which make the condition even more deadly.
"Identifying IPF endotypes could allow a degree of personalised patient management."
In this study, researchers used IPF CT scans* from two populations of approximately 145 patients. A radiologist was asked to identify those which also had PPFE and identified extensive PPFE in 31 out of 287 (or 11%) cases.
The research team then applied a computerised imaging tool, developed by Dr Eyjolfur Gudmundsson at CMIC, UCL to identify PPFE on a subset of scans from one of the populations. The algorithm was subsequently evaluated on scans separately in the two patient populations and concluded that 87 of the 287 scans (or 30%) had clinically important PPFE, a threefold increase on the radiologist's assessment.
The authors say the significant numbers of IPF patient scans, which also had PPFE, shows this is a feature, characteristic of a IPF disease subgroup/endotype.
Dr Jacob said: "Using this computerised tool we have identified the first endotype in IPF occurring in about 30% of IPF patients.
"The PPFE identified adversely affects survival and appears to occur and progress separate to the fibrosis in the patients' lungs."
The research team also retrospectively matched the IPF images which had PPFE, against patient records, and concluded the presence of PPFE is associated with faster clinical deterioration and earlier death in IPF patients.
Dr Jacob added: "Our research suggests that PPFE is a previously under-reported disease process that occurs separate to lung fibrosis but which is associated with an increased risk of death in IPF.
"Our computer algorithm identifies patients with the PPFE endotype of IPF which may inform patient selection in IPF drug trials and clinical management of disease. Our algorithm should be able to measure PPFE worsening over time in patients with IPF."
This research was funded by the Wellcome Trust and the NIHR UCLH Biomedical Research Centre.
*CT scans were obtained from IPF patient cohorts at University Hospital Southampton NHS Foundation Trust, Ege University Hospital, Izmir, Turkey and St Antonius Hospital, Nieuwegein, Netherlands.





