PHILADELPHIA – Rare inherited mutations in the body's master regulator of the DNA repair system – the TP53 gene – can leave people at a higher risk of developing multiple types of cancer over the course of their lives. Now, for the first time, a team led by researchers in the Basser Center for BRCA at the Abramson Cancer Center of the University of Pennsylvania details the potential implications of a lower risk TP53 mutation, including an association with a specific type of Li-Fraumeni syndrome (LFS), an inherited predisposition to a wide range of cancers. The researchers published their findings today in Cancer Research, a journal of the American Association for Cancer Research.
Mutations in the TP53 gene are the most commonly acquired mutations in cancer. The p53 protein, made by the TP53 gene, normally acts as the supervisor in the cell as the body tries to repair damaged DNA. Different mutations can determine how well or how poorly that supervisor is able to direct the response. The more defective the mutation, the greater the risk. When TP53 mutations are inherited, they cause LFS, a disease that leaves people with a 90 percent chance of developing cancer in their lifetime. These commonly include soft tissue and bone sarcomas, breast and brain cancer, adrenocortical tumors, and leukemia, and patients undergo frequent screening starting as infants to look for signs of disease, given the high risk of childhood cancers that continues throughout their lives. There are currently no therapies that target the p53 pathway.
"Due to the wide variety of disease types associated with inherited TP53 mutations and the early age of cancer diagnoses, cancer screening is exceptionally aggressive. However we do not yet know if all mutations require the same high level of screening," said the study's senior author Kara N. Maxwell, MD, PhD, an assistant professor of Hematology-Oncology and Genetics in the Perelman School of Medicine at the University of Pennsylvania and a member of the Abramson Cancer Center and the Basser Center for BRCA. "It is therefore critical to study the specifics of individual TP53 mutations so we can understand how best to screen people who carry lower risk mutations."
The study's co-first authors are Jacquelyn Powers, MS, LCGC, a genetic counselor in the Basser Center for BRCA, and Emilia Modolo Pinto, PhD, an associate scientist at St. Jude Children's Research Hospital.
For this study, researchers genetically sequenced multiple members of eight different families, then combined those data into a study cohort, along with data of carriers from two genetic testing cohorts. They found this newly identified mutation confers a risk of childhood cancer in some families, but only later onset cancers in others, and likely at a lower than 90 percent lifetime risk.
The St. Jude team, as well as researchers from The Wistar Institute, were critical to the next stage of the research.
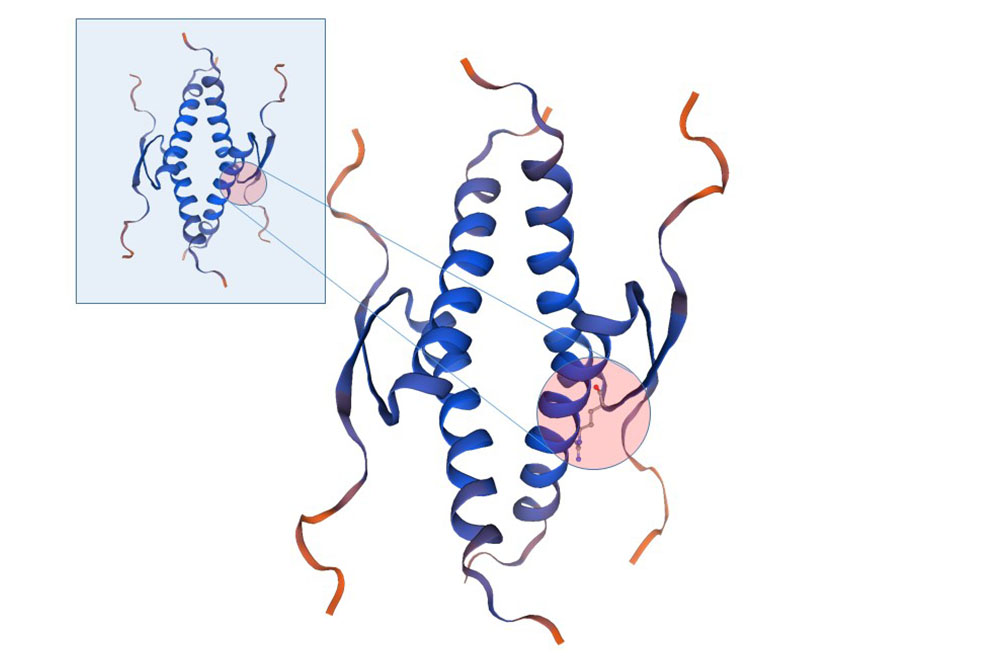
The St. Jude team modeled the TP53 mutation to try to figure out what effect it had on the protein's structure. In addition, working closely with the Penn team, the St. Jude team also determined that there is an inherited set of genetic material shared among people who have this mutation, suggesting it's what's called a founder mutation – a mutation that tracks within one ethnicity. In this case, that ethnicity is the Ashkenazi Jewish population.
"By using the same model that led us to determine a founder mutation widespread in Brazilian individuals we have determined this new one in the Ashkenazi Jewish population," Pinto said.
A team from The Wistar Institute studied the consequences of the new mutation on the function of the p53 protein in cells and showed it affects the expression levels of multiple p53 target genes, suggesting this might play a role in transformation and cancer formation.
"By identifying and understanding this Ashkenazi variant of p53, our goal is to help people who have genetic variants of this critical gene to better understand their cancer risk, and eventually to assist the development of new specific treatments that will reduce the burden of cancer on this population," said Maureen E. Murphy, PhD, Ira Brind Professor and program leader of the Molecular & Cellular Oncogenesis Program of the Wistar Cancer Center.
The findings raise questions about how to appropriately screen patients for this mutation and whether the standard process of full-body scans for LFS patients should be modified for this group, since their risk profile is different than those with classic LFS.
Maxwell and her team are part of a group working on developing liquid biopsy techniques – a blood test that would be able to improve detection. They say they're hopeful this study will help inform future liquid biopsy work.
This study was supported by the National Institutes of Health Grants (K08CA215312, P30CA016520, R01CA102184, K99CA241367, K08CA234394, KL2TR00187903, R01CA242218, RC4CA153828, R01CA225662), the Basser Center for BRCA, the Breast Cancer Research Foundation, the Burroughs Wellcome Foundation, the International Pediatric Adrenocortical Tumor Registry, and ALSAC – the fundraising and awareness organization of St. Jude.





